Pakar perubatan artikel itu

Penerbitan baru
Sindrom Pierre Robin
Ulasan terakhir: 04.07.2025

Semua kandungan iLive disemak secara perubatan atau fakta diperiksa untuk memastikan ketepatan faktual sebanyak mungkin.
Kami mempunyai garis panduan sumber yang ketat dan hanya memautkan ke tapak media yang bereputasi, institusi penyelidikan akademik dan, apabila mungkin, dikaji semula kajian secara medis. Perhatikan bahawa nombor dalam kurungan ([1], [2], dan lain-lain) boleh diklik pautan ke kajian ini.
Jika anda merasakan bahawa mana-mana kandungan kami tidak tepat, ketinggalan zaman, atau tidak dipersoalkan, sila pilih dan tekan Ctrl + Enter.

Sindrom Pierre Robin, juga dikenali dalam perubatan sebagai anomali Robin, adalah patologi kongenital perkembangan bahagian rahang muka. Penyakit ini menerima namanya sebagai penghormatan kepada doktor gigi Perancis P. Robin, yang pertama kali menerangkan semua tanda-tandanya. Lannelongue dan Menard pertama kali menggambarkan sindrom Pierre Robin pada tahun 1891 dalam laporan mereka mengenai dua pesakit dengan micrognathia, lelangit sumbing dan retroglossoptosis. Pada tahun 1926, Pierre-Robin menerbitkan satu kes penyakit pada bayi dengan tanda-tanda sindrom klasik. Sehingga tahun 1974, triad tanda dikenali sebagai sindrom Robin-Pierre. Walau bagaimanapun, sindrom ini kini digunakan untuk menggambarkan kecacatan dengan kehadiran serentak pelbagai anomali.
Epidemiologi
Ia adalah kecacatan kongenital heterogen yang mempunyai kelaziman 1 dalam 8,500 kelahiran hidup. Nisbah lelaki kepada perempuan ialah 1:1, kecuali untuk bentuk berkait-X.
Di kalangan pesakit ini, 50% bayi mempunyai lelangit lembut sumbing yang tidak lengkap, selebihnya dilahirkan dengan lelangit melengkung dan luar biasa tinggi, tetapi tanpa sumbing.
Punca Sindrom Pierre Robin
Kemungkinan pewarisan resesif autosomal penyakit ini dipertimbangkan. Terdapat dua jenis sindrom bergantung kepada etiologi: terpencil dan ditentukan secara genetik. Jenis terpencil berkembang kerana mampatan bahagian bawah rahang semasa perkembangan embrio. Mampatan boleh berkembang disebabkan oleh:
- Kehadiran anjing laut setempat dalam rahim (sista, parut, tumor).
- Kehamilan berganda.
Juga, perkembangan rahang dalam janin boleh terganggu oleh:
- Jangkitan virus yang dihidapi ibu mengandung semasa mengandung.
- Gangguan neurotropik.
- Jumlah asid folik yang tidak mencukupi dalam badan wanita hamil.
Patogenesis
Sindrom Pierre Robin disebabkan oleh gangguan embrio yang disebabkan oleh pelbagai jenis patologi dalam tempoh pranatal.
Terdapat tiga teori patofisiologi yang mungkin menjelaskan kejadian sindrom Pierre Robin.
Teori Mekanikal: Teori ini adalah yang paling mungkin. Kurang pembangunan radas mandibular berlaku antara minggu ke-7 dan ke-11 kehamilan. Kedudukan lidah yang tinggi dalam rongga mulut membawa kepada pembentukan celah di lelangit, yang menyebabkan vena cava tidak menutup. Teori ini menerangkan celah klasik berbentuk U terbalik dan ketiadaan bibir sumbing yang berkaitan. Oligohydramnios mungkin memainkan peranan dalam etiologi, kerana ketiadaan cecair amniotik boleh menyebabkan ubah bentuk dagu dan pemampatan seterusnya pada lidah antara vena kava.
Teori neurologi: Kelewatan dalam perkembangan neurologi telah diperhatikan dengan elektromiografi otot-otot uvula dan lajur pharyngeal, dan rasa akibat kelewatan konduksi dalam saraf hypoglossal.
Teori disneuroregulasi rhombencephalon: Teori ini berdasarkan gangguan perkembangan rhombencephalon semasa ontogenesis.
Perkembangan bahagian bawah rahang kanak-kanak yang tidak mencukupi menyebabkan rongga mulut berkurangan dengan ketara. Ini, seterusnya, menyebabkan apa yang dipanggil pseudomacroglossia, iaitu lidah disesarkan ke belakang dinding pharyngeal. Patologi ini membawa kepada perkembangan halangan saluran udara.
Selagi bayi menangis atau bergerak, saluran pernafasan tetap bersih, tetapi sebaik sahaja bayi tertidur, halangan berlaku lagi.
Disebabkan gangguan pernafasan, proses penyusuan bayi amat sukar. Pada masa ini, halangan saluran pernafasan hampir selalu berlaku. Sekiranya tiada pembetulan perubatan digunakan, patologi seperti itu boleh menyebabkan keletihan yang teruk pada seluruh badan dan juga kematian.
Gejala Sindrom Pierre Robin
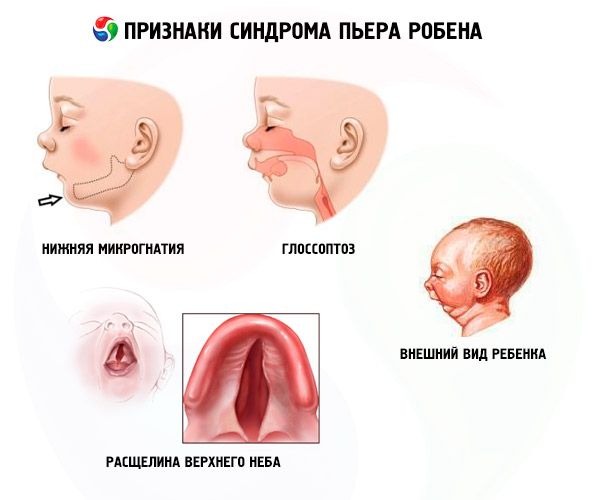
Penyakit ini dicirikan oleh tiga gejala utama:
- Micrognathia yang lebih rendah (kurang pembangunan rahang bawah, berlaku dalam 91.7% kes penyakit). Ia dicirikan oleh penarikan balik gerbang pergigian bawah sebanyak 10-12 mm di belakang gerbang atas. Rahang bawah mempunyai badan kecil, sudut tumpul. Kanak-kanak mencapai perkembangan normal pada usia 5-6 tahun.
- Glossoptosis (penarikan balik lidah kerana perkembangannya yang tidak mencukupi, diperhatikan dalam 70-85% kes).
- Macroglossia dan ankyloglossia adalah gejala yang agak jarang berlaku, diperhatikan dalam 10-15% kes.
- Sebuah retakan muncul di langit.
- Bradipnea dan dyspnea.
- Sianosis ringan.
- Asfiksia, yang paling kerap berlaku semasa percubaan untuk memberi makan kepada bayi.
- Menelan adalah mustahil atau sangat sukar.
- Rasa nak muntah.
- Anomali aurikular dalam 75% kes.
- Kehilangan pendengaran konduktif berlaku pada 60% pesakit, manakala atresia saluran pendengaran luaran berlaku hanya pada 5% pesakit, pneumatisasi tidak mencukupi rongga mastoid tulang temporal.
- Anomali telinga dalam (aplasia saluran separuh bulatan sisi, saluran air vestibular yang besar, kehilangan sel rambut koklea).
- Kecacatan hidung jarang berlaku dan terdiri terutamanya daripada anomali akar hidung.
- Kecacatan gigi berlaku dalam 30% kes. Laryngomalacia dan kekurangan velopharyngeal berlaku pada kira-kira 10-15% pesakit dengan sindrom Pierre Robin.
Ciri sistemik sindrom Pierre Robin
Anomali perkembangan sistemik diterangkan dalam 10-85% kes berdaftar.
Keabnormalan mata berlaku pada 10-30% pesakit. Mereka mungkin termasuk: hiperopia, miopia, astigmatisme, sklerosis kornea dan stenosis saluran nasolakrimal.
Patologi kardiovaskular: murmur jantung benigna, stenosis arteri pulmonari, duktus arteriosus paten, tingkap bujur, kecacatan septum atrium dan hipertensi pulmonari. Kelaziman mereka berbeza dari 5-58%.
Anomali yang berkaitan dengan sistem muskuloskeletal (70-80% daripada kes): syndactyly, dysplastic phalanges, polydactyly, clinodactyly, hypermobility sendi dan oligodactyly pada anggota atas. Anomali anggota bawah: anomali kaki (kaki kelab, adduksi metatarsal), kecacatan femoral (valgus atau varus pelvis, femur pendek), anomali pinggul (dislokasi kongenital, kontraktur), anomali sendi lutut (GENU VALGUS, synchondrosis). Kecacatan tulang belakang: scoliosis, kyphosis, lordosis, displasia vertebra, agenesis sakrum dan sinus coccygeal.
Patologi sistem saraf pusat: epilepsi, kelewatan dalam perkembangan sistem saraf, hidrosefalus. Kekerapan kecacatan CNS adalah kira-kira 50%.
Anomali genitouriner: buah zakar tidak turun (25%), hidronefrosis (15%), dan hidrokel (10%).
Sindrom dan keadaan yang berkaitan: Sindrom Stickler, sindrom trisomi 11q, trisomi 18, sindrom pemadaman 4q, artropati reumatoid, hipokondroplasia, sindrom Moebius.
Tahap
Terdapat tiga peringkat keterukan penyakit, yang bergantung kepada keadaan saluran pernafasan kanak-kanak:
- Ringan - terdapat masalah kecil dengan pemakanan, tetapi pernafasan hampir tidak sukar. Rawatan dijalankan secara pesakit luar.
- Sederhana - pernafasan adalah sederhana sukar, memberi makan kanak-kanak adalah sederhana sukar. Rawatan dijalankan di hospital.
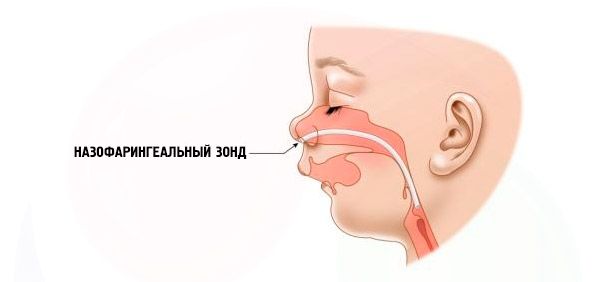
- Teruk - pernafasan sangat sukar, kanak-kanak tidak boleh diberi makan secara normal. Ia perlu menggunakan peranti khas (probe intranasal).
Komplikasi dan akibatnya
Gabungan micrognathia dan glossoptosis boleh membawa kepada komplikasi pernafasan yang teruk dan masalah semasa memberi makan kepada kanak-kanak.
Sindrom Pierre Robin menyebabkan komplikasi berikut:
- Pernafasan Stridose disebabkan oleh halangan saluran pernafasan. Laryngomalacia atau asfiksia tidur.
- Perkembangan psikomotor kanak-kanak jauh ketinggalan berbanding rakan sebayanya.
- Perkembangan fizikal juga ketinggalan.
- Pertuturan pesakit terganggu.
- Jangkitan telinga yang kerap menjadi kronik dan membawa kepada gangguan pendengaran.
- Sindrom apnea tidur obstruktif, kejadian kematian dalam tidur berbeza-beza dalam 14-91% kes.
- Masalah dengan gigi.
Diagnostik Sindrom Pierre Robin
Diagnosis sindrom Pierre Robin tidak sukar. Ia berdasarkan manifestasi klinikal. Untuk mengecualikan patologi lain, sangat penting untuk berunding dengan ahli genetik.
Kanak-kanak yang mengalami anomali kongenital Robin mengalami masalah pernafasan sejak lahir kerana lidah sentiasa tenggelam ke belakang. Bayi resah, kulitnya kebiruan, semput keluar dari dada apabila menarik nafas. Tercekik mungkin berlaku semasa penyusuan. Diagnosis juga boleh dibuat oleh penampilan luar biasa kanak-kanak itu - "muka burung". Selalunya, pesakit mengalami kecacatan lain: miopia, katarak, patologi sistem genitouriner, patologi jantung, anomali dalam perkembangan tulang belakang.
Berdasarkan manifestasi klinikal ini, tidak sukar bagi pakar untuk membuat diagnosis yang betul.
Siapa yang hendak dihubungi?
Rawatan Sindrom Pierre Robin
Rawatan dijalankan sejurus selepas kelahiran seorang kanak-kanak dengan sindrom Pierre Robin. Sekiranya penyakit itu ringan, maka untuk memperbaiki keadaan pesakit adalah perlu untuk sentiasa memegang anak secara menegak atau berbaring di atas perut. Kepala bayi hendaklah dicondongkan ke dada. Semasa memberi makan, tidak disyorkan untuk memegang kanak-kanak dalam kedudukan mendatar supaya makanan tidak masuk ke saluran pernafasan.
Sekiranya keterbelakangan rahang bawah agak ketara, campur tangan pembedahan digunakan untuk membawa lidah yang ditarik balik ke kedudukan fisiologi yang normal. Dalam kes yang teruk, lidah ditarik ke atas dan dipasang pada bibir bawah. Dalam kes yang sangat teruk, trakeostomi, glossopeksi, dan gangguan osteogenesis rahang bawah mesti dilakukan.
Rawatan konservatif juga digunakan.
Ubat-ubatan
Fenobarbital. Pil tidur dan ubat penenang, mempunyai kesan anticonvulsant. Setiap tablet mengandungi 100 ml phenobarbital. Dos adalah individu, kerana ia bergantung kepada keparahan penyakit dan keadaan kanak-kanak. Ubat ini dilarang untuk pesakit yang mengalami kegagalan hati, hyperkinesis, anemia, myasthenia, porfiria, diabetes mellitus, kemurungan, dan intoleransi terhadap komponen. Gejala berikut mungkin berlaku apabila mengambilnya: pening, asthenia, halusinasi, agranulositosis, loya, tekanan darah rendah, dan alahan.
Clonazepam. Ubat yang ditetapkan untuk rawatan epilepsi. Ubat ini mengandungi bahan aktif clonazepam, yang merupakan derivatif benzodiazepine. Ia mempunyai kesan anticonvulsant, anxiolytic dan relaxant otot. Dos ditentukan oleh doktor yang hadir, tetapi tidak boleh melebihi maksimum - 250 mcg sehari. Jangan ambil sekiranya insomnia, hipertonia otot, pergolakan psikomotor, gangguan panik. Gejala berikut adalah mungkin apabila mengambil: kelesuan, loya, dysmenorrhea, sakit kepala, leukopenia, pengekalan kencing atau inkontinensia, alopecia, alahan.
Sibazon. Terdapat dalam bentuk penyelesaian dan tablet rektum. Bahan aktifnya ialah derivatif benzodiazepine (sibazon). Ia mempunyai kesan sedatif, anxiolytic, anticonvulsant. Dos adalah individu. Pesakit dengan hiperkapnia kronik, myasthenia, intoleransi benzodiazepine dilarang mengambil ubat. Apabila menggunakan ubat, gejala berikut mungkin berkembang: loya, sembelit, sakit kepala, pening, cegukan, inkontinensia kencing, alahan.
Cortexin lyophilisate. Ubat dengan kesan nootropik. Ubat ini mengandungi kompleks pecahan polipeptida larut air dan glisin. Dos adalah individu dan ditetapkan oleh doktor yang merawat mengikut keadaan pesakit. Pesakit dengan intoleransi terhadap cortexin dilarang mengambil ubat. Dadah boleh menyebabkan reaksi alahan.
Rawatan fisioterapi
Biasanya, dalam peringkat ringan sindrom, terapi posisi digunakan, di mana kanak-kanak diletakkan di atas perutnya dalam kedudukan tegak sehingga graviti memaksa rahang bawah untuk membesar dengan betul.
Rawatan pembedahan
Rawatan pembedahan digunakan terutamanya untuk membetulkan glossoptosis. Terdapat beberapa kaedah:
- Sokongan lidah dengan benang perak. Benang disalurkan melalui bahagian bawah gusi dan bibir bawah. Kaedah itu dipanggil Douglas.
- Kaedah Duhamel - benang perak tebal disalurkan melalui pangkal lidah pesakit dan kedua-dua pipi. Gunakan tidak lebih daripada tiga puluh hari.
- Peranti ortopedik untuk sambungan dan penetapan lidah.
- Pada usia satu tahun, pembedahan untuk membetulkan lelangit sumbing boleh dilakukan.
Ramalan
Prognosis dan perjalanan penyakit adalah teruk. Selalunya, kematian berlaku pada hari-hari pertama kehidupan pada tahap sederhana dan teruk penyakit (penyebabnya adalah asfiksia). Juga, risiko kematian pada tahun pertama agak tinggi disebabkan oleh banyak jangkitan.
Bagi pesakit yang berumur lebih dari dua tahun, prognosis adalah baik.
 [ 36 ]
[ 36 ]