Pakar perubatan artikel itu

Penerbitan baru
Kebodohan Amaurotik
Last reviewed: 04.07.2025

Semua kandungan iLive disemak secara perubatan atau fakta diperiksa untuk memastikan ketepatan faktual sebanyak mungkin.
Kami mempunyai garis panduan sumber yang ketat dan hanya memautkan ke tapak media yang bereputasi, institusi penyelidikan akademik dan, apabila mungkin, dikaji semula kajian secara medis. Perhatikan bahawa nombor dalam kurungan ([1], [2], dan lain-lain) boleh diklik pautan ke kajian ini.
Jika anda merasakan bahawa mana-mana kandungan kami tidak tepat, ketinggalan zaman, atau tidak dipersoalkan, sila pilih dan tekan Ctrl + Enter.

Amaurotic idiocy adalah penyakit progresif yang jarang berlaku. Ia dicirikan oleh penurunan penglihatan secara beransur-ansur untuk melengkapkan kebutaan dan kemerosotan kecerdasan sehingga kebodohan berlaku. Akibatnya, pesakit mengalami marasmus yang mendalam dengan hasil yang membawa maut. Penyakit ini pertama kali diterangkan oleh pakar oftalmologi Dr Tau lebih 130 tahun dahulu. Tau mencatatkan transformasi khas fundus. Lebih daripada 500 kes penyakit itu telah diterangkan.
 [ 1 ], [ 2 ], [ 3 ], [ 4 ], [ 5 ], [ 6 ], [ 7 ], [ 8 ], [ 9 ], [ 10 ]
[ 1 ], [ 2 ], [ 3 ], [ 4 ], [ 5 ], [ 6 ], [ 7 ], [ 8 ], [ 9 ], [ 10 ]
Epidemiologi
Epidemiologi penyakit ini mencatatkan sifat kekeluargaan. Dalam keluarga di mana terdapat pesakit dengan kebodohan amaurotik, perkahwinan seangkatan adalah berbahaya untuk sebab yang sama. Turut berisiko ialah orang yang berasal dari Kanada-Perancis atau Yahudi.
[ 11 ], [ 12 ], [ 13 ], [ 14 ], [ 15 ], [ 16 ], [ 17 ], [ 18 ]
Punca kebodohan amaurotik
Walaupun terdapat banyak data yang dikumpul mengenai penyakit itu, para saintis pada masa ini terus mencari jawapan kepada banyak soalan mengenai punca, patogenesis dan juga manifestasi kebodohan amaurotik.
Terdapat cadangan bahawa penyakit itu adalah keturunan. Jenis pewarisan adalah resesif autosomal. Selalunya, cerebellum dan lobus oksipital hemisfera serebrum terjejas, dengan akibat dan komplikasi yang teruk untuk seluruh badan: atrofi saraf optik, gentian saraf boleh kehilangan membrannya, dan sambungan antara sel saraf boleh hancur.
Kebanyakan pakar mengakui bahawa tanda-tanda klinikal penyakit ini boleh agak berbeza-beza dan berkait dengan usia di mana kebodohan amaurotik mula berkembang pada pesakit.
Semasa kajian punca penyakit itu, corak tertentu diperhatikan: penyakit ini sering memberi kesan kepada kanak-kanak dari keluarga yang sama, itulah sebabnya nama "kebodohan amaurotik keluarga" digunakan. Menurut kajian, keputusan yang diterbitkan ketika mereka baru mula mengkaji penyakit itu, daripada 64 kes kebodohan amaurotik, 37 ditemui dalam 13 keluarga (setiap keluarga mempunyai 2-5 anak yang sakit). Perlu diperhatikan bahawa dalam keluarga sedemikian, orang yang sakit mempunyai saudara lelaki dan perempuan yang benar-benar sihat. Pada masa kini, dipercayai bahawa faktor pewarisan resesif memainkan peranan yang besar dalam perkembangan penyakit ini. Oleh itu, adalah mungkin untuk menjelaskan kekerapan berlakunya kes penyakit dalam keluarga yang sama. Apabila menganalisis faktor keturunan sebagai punca kebodohan amaurotik, seseorang tidak boleh mengehadkan diri kepada kehadiran tanda-tanda yang dinyatakan secara klinikal dalam keluarga pesakit (kedua-duanya dalam garis menaik dan sisi), tetapi juga mengambil kira yang asas, sebagai contoh, penyimpangan ciri dalam fungsi alat visual (koroiditis keluarga, distrofi pigmen dan lain-lain. retina).
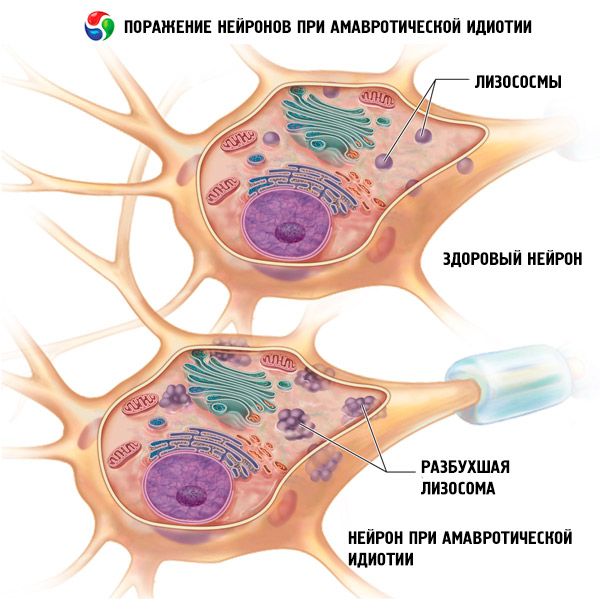
Gejala kebodohan amaurotik
Tanda-tanda pertama bentuk kongenital muncul pada hari-hari pertama atau minggu-minggu kehidupan. Bayi itu dilahirkan dengan hidrosefalus atau microcephaly, mengalami sawan, lumpuh, dan disfungsi pernafasan. Kanak-kanak itu mati selepas beberapa bulan.
Tahap
Bentuk bayi berkembang dari 4-6 bulan. Bentuk idiopathi amaurotik ini dicirikan oleh sifat kekeluargaan. Penglihatan merosot dengan cepat: bayi tidak dapat membetulkan pandangannya, tidak memerhati objek. Apa yang dipanggil "lubang ceri" muncul pada fundus - bintik kemerahan di kawasan makula, yang dikelilingi oleh rim kelabu-putih. Kemudian saraf optik atrofi, dan kanak-kanak itu benar-benar kehilangan keupayaan untuk melihat. Orientasi, refleks pelindung, serta keupayaan untuk bergerak, secara beransur-ansur hilang. Pesakit bertindak balas dengan kuat terhadap rangsangan bunyi - mereka tersentak dari bunyi yang senyap untuk orang yang sihat, sawan mungkin diperhatikan kerana peningkatan nada otot. Pada peringkat akhir penyakit, atrofi umum, keletihan badan dan peningkatan nada semua otot extensor berkembang. Prognosis penyakit ini juga mengecewakan: pesakit mati satu setengah hingga dua tahun selepas permulaan penyakit.
Bentuk zaman kanak-kanak lewat bermula pada 3-4 tahun. Penyakit progresif bergantian dengan peringkat remisi. Kehilangan kecerdasan secara beransur-ansur disertai dengan sawan, gangguan koordinasi, dan gangguan ekstrapiramidal. Bentuk ini juga dicirikan oleh atrofi saraf optik. Kematian berlaku 6-8 tahun selepas bermulanya kebodohan amaurotik.
Bentuk juvana mula menampakkan dirinya pada 6-10 tahun. Kebodohan amaurotik Spielmeyer berkembang kurang cepat. Perubahan dalam fundus bertepatan dengan manifestasi distrofi retina pigmen. Penglihatan pesakit secara beransur-ansur menurun, begitu juga dengan kecerdasan. Fungsi motor terjejas boleh menampakkan diri dengan cara yang berbeza dan tidak tetap: kelumpuhan lengan dan kaki yang tidak begitu ketara, gangguan extrapyramidal dan bulbar berlaku. Penyakit ini membawa kepada kematian 10-25 tahun selepas perkembangan tanda-tanda pertama.
Bentuk lewat berlaku sangat jarang dan berkembang sangat perlahan. Keadaan mental pesakit berubah (seperti sindrom mental organik), atrofi saraf optik dan distrofi pigmen retina diperhatikan. Peringkat akhir dicirikan oleh kelumpuhan dan sindrom epileptiform. Pesakit mati 10-15 tahun selepas permulaan penyakit.
[ 38 ], [ 39 ], [ 40 ], [ 41 ], [ 42 ], [ 43 ], [ 44 ], [ 45 ]
Borang
Terdapat empat jenis kebodohan amaurotik:
- Tay-Sachs (menjejaskan pada usia awal);
- Jansky-Bilynovsky (muncul pada kanak-kanak pada usia lanjut);
- Sindrom Spielmeyer-Vogt (berlaku pada remaja);
- Kufsa (bentuk akhir).
Sesetengah saintis juga membezakan jenis Norman-Wood kongenital secara berasingan.
Setiap jenis penyakit mempunyai set manifestasi klinikalnya sendiri, tetapi semuanya disatukan oleh sebab biasa, gambaran klinikal, asas anatomi dan patogenesis.
[ 46 ], [ 47 ], [ 48 ], [ 49 ], [ 50 ], [ 51 ], [ 52 ], [ 53 ], [ 54 ]
Diagnostik kebodohan amaurotik
Amaurotic idiocy disebabkan oleh gangguan metabolisme lipid, akibatnya produk perantaraan metabolisme lipid, sphingomyelin, didepositkan dalam pelbagai sel badan. Lokasi dan komposisi deposit menentukan perkembangan gambaran klinikal penyakit tertentu.
[ 55 ], [ 56 ], [ 57 ], [ 58 ], [ 59 ], [ 60 ], [ 61 ], [ 62 ], [ 63 ]
Bagaimana untuk memeriksa?
Diagnosis pembezaan
Diagnosis pembezaan kebodohan amaurotik adalah berdasarkan gambaran klinikal tertentu dan patologi ciri fundus.
Bentuk awal mempunyai simptom yang serupa dengan penyakit Landing, sejenis mucopolysaccharidosis. Penyakit pendaratan berkembang dari bulan pertama selepas kelahiran dan membawa kepada kematian selepas 2-3 tahun. "Lubang ceri" pada fundus muncul dalam 1/5 kes, perubahan degeneratif dalam retina dan persepsi bunyi yang terdistorsi (hiperkusi) secara praktikalnya tidak menjadi cirinya, tetapi pembesaran limpa dan hati secara serentak, gangguan mental dan gangguan pergerakan diperhatikan.
Bentuk juvana kadangkala bertindih dengan manifestasi sindrom Lawrence-Moon-Biedl. Untuk membezakan penyakit ini, adalah perlu untuk memberi perhatian kepada manifestasi lain mereka. Sindrom Lawrence-Moon-Biedl dicirikan oleh penambahan berat badan yang cepat, ubah bentuk anggota badan yang dicirikan oleh kehadiran jari atau jari kaki tambahan, gangguan vegetatif-trofik yang ketara dan ketiadaan gangguan fungsi motor.
Kepelbagaian gejala kebodohan amaurotik lewat merumitkan diagnosis semasa hidup. Manifestasinya adalah serupa dengan ataxia Friedreich, multiple sclerosis, penyakit Alzheimer, penyakit Pick, lumpuh progresif, dan juga skizofrenia.
Sesetengah penulis menegaskan bahawa diagnosis penyakit ini, terutamanya apabila manifestasi klinikal kabur, boleh dipercayai hanya selepas kematian, berdasarkan analisis keabnormalan histologi sistem saraf.
Siapa yang hendak dihubungi?
Rawatan kebodohan amaurotik
Tiada rawatan yang rasional dan berkesan. Pada masa kini, terapi untuk kebodohan amaurotik bertujuan secara eksklusif untuk melegakan gejala. Sedatif, nootropik, antikonvulsan dan tonik am digunakan.
Untuk mengaktifkan peredaran darah dan proses metabolik di otak, glisin, elkar, cerebrolysin, asid glutamat, dan pantogam ditetapkan.
Untuk melegakan sindrom konvulsi, diphenin atau carmazepine ditetapkan.
Keputusan positif boleh dicapai dengan menggunakan ekstrak tisu, pemindahan darah atau plasma.
Pencegahan
Kekurangan terapi yang berkesan untuk kebodohan amaurotik memaksa kita untuk memberi perhatian kepada pencegahan. Sudah ada kaedah yang membolehkan kita mengenal pasti pembawa heterozigot gen patologi dan kaedah untuk mendiagnosis kebodohan amaurotik semasa kehamilan. Diagnostik antenatal penyakit ini terdiri daripada menganalisis aktiviti hexosaminidase A dalam cecair amniotik. Sekiranya aktiviti enzim berkurangan dikesan, disyorkan untuk menamatkan kehamilan. Ibu bapa kepada anak yang sakit dinasihatkan untuk berhenti melahirkan anak.
Использованная литература