Pakar perubatan artikel itu

Penerbitan baru
penyakit Huntington
Ulasan terakhir: 05.07.2025

Semua kandungan iLive disemak secara perubatan atau fakta diperiksa untuk memastikan ketepatan faktual sebanyak mungkin.
Kami mempunyai garis panduan sumber yang ketat dan hanya memautkan ke tapak media yang bereputasi, institusi penyelidikan akademik dan, apabila mungkin, dikaji semula kajian secara medis. Perhatikan bahawa nombor dalam kurungan ([1], [2], dan lain-lain) boleh diklik pautan ke kajian ini.
Jika anda merasakan bahawa mana-mana kandungan kami tidak tepat, ketinggalan zaman, atau tidak dipersoalkan, sila pilih dan tekan Ctrl + Enter.
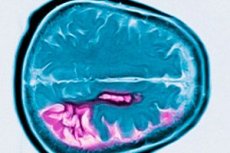
Penyakit Huntington adalah gangguan neurodegeneratif dominan autosomal yang dicirikan oleh penurunan kognitif yang progresif, pergerakan tidak disengajakan, dan koordinasi motor terjejas bermula pada usia pertengahan. Diagnosis disahkan oleh ujian genetik. Rawatan adalah terutamanya simptomatik. Ujian genetik mungkin disyorkan untuk saudara darah. George Huntington mula-mula menggambarkan keadaan itu pada tahun 1872, selepas mengkaji kes keluarga di penduduk Long Island.
Kelaziman penyakit Huntington adalah kira-kira 10 kes bagi setiap 100,000 penduduk, dan memandangkan kemunculannya lewat, kira-kira 30 orang daripada 100,000 mempunyai risiko 50% untuk membangunkannya sepanjang hayat mereka. Walaupun penyakit ini paling kerap muncul di antara umur 35 dan 40 tahun, julat umur permulaan adalah agak luas, dengan permulaan terawal adalah pada usia 3 tahun dan yang terkini pada umur 90 tahun. Walaupun penyakit ini pada asalnya dianggap mempunyai penetrasi 100%, ini kini dipercayai tidak selalu berlaku. Pada individu yang mewarisi gen penyakit daripada bapa mereka, penyakit itu menunjukkan dirinya secara purata 3 tahun lebih awal daripada mereka yang mewarisi gen patologi daripada ibu mereka. Dalam kira-kira 80% pesakit yang mewarisi gen patologi daripada bapa mereka, penyakit itu menunjukkan dirinya sebelum umur 20 tahun. Fenomena manifestasi awal kecacatan genetik dalam keturunan dipanggil jangkaan.
 [ 1 ]
[ 1 ]
Apa yang menyebabkan penyakit Huntington?
Penyakit Huntington tidak mempunyai keutamaan jantina. Atrofi nukleus caudate ditunjukkan, di mana neuron kecil merosot dan tahap neurotransmitter - asid gamma-aminobutyric (GABA) dan bahan P - jatuh.
Gen mutan dengan peningkatan bilangan ("pengembangan") urutan DNA CAG (cysteine-alanine-glycine) yang mengekod glutamin asid amino bertanggungjawab untuk perkembangan penyakit Huntington. Produk gen ini, huntingtin protein yang besar, mengandungi jumlah residu polyglutamine yang berlebihan, yang membawa kepada penyakit ini melalui mekanisme yang tidak diketahui. Semakin banyak CAG berulang, semakin awal penyakit itu muncul dan semakin teruk perjalanannya. Dari generasi ke generasi, bilangan ulangan boleh meningkat, yang dari masa ke masa membawa kepada keterukan fenotip keluarga.
Walaupun minat yang besar dalam perubahan genetik dan biokimia dalam penyakit Parkinson, pencarian gen untuk penyakit itu tidak berjaya sehingga akhir 1970-an. Pada masa itu, Nancy Wexler dan Allan Tobin menganjurkan bengkel yang ditaja oleh Yayasan Penyakit Keturunan untuk membincangkan strategi mencari gen untuk penyakit Huntington. David Housman, David Botstein, dan Ray White, yang menghadiri mesyuarat itu, mencadangkan bahawa teknik DNA rekombinan yang baru dibangunkan mungkin membantu mencapai matlamat ini. Tugas utama dalam projek itu adalah untuk mencari keluarga besar dengan banyak generasi penyakit Huntington untuk mendapatkan sampel DNA. Pada tahun 1979, projek bersama saintis dari Venezuela dan Amerika Syarikat telah dilancarkan untuk memeriksa sebuah keluarga besar dengan penyakit Huntington yang tinggal di pantai Tasik Maracheibo (Venezuela). Pada tahun 1983, gen penyakit Huntington telah dilokalkan pada penghujung lengan pendek kromosom 4 (Gusella et al., 1983), dan sedekad kemudiannya ia telah mendedahkan bahawa mutasi gen ini terdiri daripada peningkatan bilangan ulangan cytosine-adenine-guanine (CAG) trinucleotide Disease (Huntington's Disease 1999) Kumpulan Penyelidikan (1999). Metodologi yang dibangunkan oleh kumpulan saintifik ini kini dianggap standard untuk pengklonan kedudukan gen baru.
Walaupun gen jenis liar mempunyai regangan 10-28 ulangan CAG, bentuk mutan gen yang menyebabkan penyakit Huntington mempunyai regangan yang meningkat daripada 39 kepada lebih daripada 100 ulangan CAG. Penemuan pengembangan ulangan trinukleotida telah membantu menjelaskan banyak ciri klinikal penyakit ini. Khususnya, korelasi songsang ditemui antara umur permulaan dan panjang rantau dengan trinukleotida berulang. Jangkaan warisan bapa boleh dijelaskan oleh fakta bahawa peningkatan bilangan ulangan sering berlaku pada lelaki semasa spermatogenesis. Analisis mutasi baru telah menunjukkan bahawa ia biasanya berlaku apabila salah seorang ibu bapa, biasanya bapa, mempunyai kiraan ulangan CAG lebih tinggi daripada 28; dalam kes ini, bilangan ulangan ini meningkat pada generasi seterusnya. Kini telah ditetapkan bahawa jika bilangan ulangan tidak lebih daripada 28, ia akan dihantar secara stabil dari generasi ke generasi. Sekiranya bilangan ulangan adalah dari 29 hingga 35, maka gejala penyakit Huntington tidak muncul, tetapi apabila diteruskan kepada keturunan, panjang wilayah ini mungkin meningkat. Sekiranya bilangan ulangan adalah dari 36 hingga 39, maka dalam beberapa kes (tetapi tidak selalu) penyakit itu mungkin nyata secara klinikal (penembusan tidak lengkap), dan apabila diturunkan kepada anak, peningkatan bilangan ulangan trinukleotida adalah mungkin. Sekiranya bilangan ulangan melebihi 40, maka penyakit itu berlaku dalam hampir semua kes, dan apabila diteruskan kepada anak, pengembangan ulangan selanjutnya adalah mungkin. Sebab-sebab peningkatan bilangan ulangan masih tidak diketahui.
Patomorfologi penyakit Huntington
Penyakit Huntington dicirikan oleh kehilangan neuron terutamanya dalam nukleus caudate dan putamen, dan pada tahap tertentu juga dalam korteks dan struktur otak yang lain. Jumlah berat otak dalam penyakit Huntington dikurangkan bukan sahaja dengan penurunan bilangan neuron, tetapi juga oleh kehilangan bahan putih. Dalam korteks serebrum, sel dalam lapisan V dan VI paling terjejas. Keterukan perubahan degeneratif mikro dan makroskopik (dilaraskan untuk umur semasa kematian) berkorelasi dengan bilangan ulangan CAG. Analisis patologi terperinci tentang perubahan dalam beberapa ratus kes penyakit Huntington telah menunjukkan bahawa degenerasi striatum bermula di bahagian dorsomedial nukleus caudate dan bahagian dorsolateral putamen, dan kemudian merebak secara ventral. Kumpulan neuron yang berbeza dalam nukleus caudate dan putamen terjejas pada tahap yang berbeza. Interneuron dalam striatum kekal secara relatifnya utuh, tetapi beberapa neuron unjuran terjejas secara selektif. Dalam bentuk juvana penyakit Huntington, perubahan patomorfologi dalam striatum lebih ketara dan lebih meluas, melibatkan korteks serebrum, cerebellum, talamus, dan globus pallidus.
Perubahan neurokimia dalam penyakit Huntington
GABA. Kajian neurokimia otak pada pesakit dengan penyakit Huntington mendedahkan penurunan ketara dalam kepekatan GABA dalam striatum. Kajian seterusnya mengesahkan bahawa penyakit Huntington dikaitkan dengan penurunan bilangan neuron GABAergik dan menunjukkan bahawa kepekatan GABA dikurangkan bukan sahaja di striatum tetapi juga di zon unjurannya - segmen luaran dan dalaman globus pallidus dan substantia nigra. Dalam otak dalam penyakit Huntington, perubahan dalam reseptor GABA juga dikesan menggunakan kajian pengikatan reseptor dan hibridisasi in situ mRNA. Bilangan reseptor GABA dikurangkan secara sederhana dalam nukleus caudate dan putamen, tetapi meningkat di bahagian retikular substantia nigra dan segmen luar globus pallidus, yang mungkin disebabkan oleh hipersensitiviti denervasi.
Asetilkolin. Acetylcholine digunakan sebagai neurotransmitter oleh interneuron nonspiny besar dalam striatum. Kajian postmortem awal pada pesakit dengan penyakit Huntington menunjukkan penurunan aktiviti choline acetyltransferase (ChAT) dalam striatum, mencadangkan kehilangan neuron kolinergik. Walau bagaimanapun, berbanding dengan pengurangan ketara dalam neuron GABAergik, interneuron kolinergik agak terhindar. Oleh itu, ketumpatan neuron positif acetylcholinesterase dan aktiviti ChAT dalam striatum sebenarnya agak tinggi berbanding dengan kawalan yang dipadankan dengan umur.
Bahan P. Bahan P terkandung dalam banyak neuron berduri sederhana striatum, yang kebanyakannya mengunjur ke segmen dalaman globus pallidus dan substantia nigra dan biasanya juga mengandungi dynorphin dan GABA. Tahap bahan P dalam striatum dan pars reticularis substantia nigra berkurangan dalam penyakit Huntington. Pada peringkat terminal penyakit ini, kajian imunohistokimia telah mendedahkan pengurangan ketara dalam bilangan neuron yang mengandungi bahan P. Pada peringkat awal, neuron yang mengandungi bahan P dan mengunjurkan ke segmen dalaman globus pallidus agak terhindar, berbanding dengan neuron yang mengunjur ke pars reticularis substantia nigra.
Peptida opioid. Enkephalin terkandung dalam neuron GABAergic unjuran berduri sederhana dari laluan tidak langsung, yang mengunjur ke segmen luar globus pallidus dan membawa reseptor D2. Kajian imunohistokimia telah menunjukkan bahawa neuron yang mengandungi enkephalin yang menonjol ke segmen luar globus pallidus hilang pada awal penyakit Huntington. Sel-sel ini nampaknya mati lebih awal daripada sel yang mengandungi bahan P yang menonjol ke segmen dalaman globus pallidus.
Katekolamin. Neuron yang mengandungi amina biogenik (dopamin, serotonin) dan menonjol ke striatum terletak di bahagian padat substantia nigra, tegmentum ventral, dan nukleus raphe. Walaupun unjuran noradrenergik kepada striatum manusia adalah minimum, paras serotonin dan dopamin (setiap gram tisu) dalam striatum dinaikkan, menunjukkan pengekalan unjuran aferen ini walaupun kehilangan ketara neuron striatum itu sendiri. Neuron dopaminergik substantia nigra kekal utuh dalam kedua-dua bentuk klasik dan juvana penyakit Huntington.
Somatostatin/neuropeptide Y dan nitric oxide synthetase. Pengukuran tahap somatostatin dan neuropeptida Y dalam striatum dalam penyakit Huntington menunjukkan peningkatan 4-5 kali ganda berbanding dengan tisu biasa. Kajian imunohistokimia menunjukkan pemeliharaan mutlak interneuron striatal yang mengandungi neuropeptida Y, somatostatin dan nitric oxide synthetase. Oleh itu, neuron ini tahan terhadap proses patologi.
Asid amino pengujaan. Telah dicadangkan bahawa kematian sel terpilih dalam penyakit Huntington adalah disebabkan oleh kesan neurotoksik yang disebabkan oleh glutamat. Tahap glutamat dan asid quinolinik (neurotoksin endogen yang merupakan hasil sampingan metabolisme serotonin dan agonis reseptor glutamat) dalam striatum penyakit Huntington sedikit diubah, tetapi kajian terbaru menggunakan spektroskopi MR mendedahkan peningkatan dalam tahap glutamat dalam vivo. Tahap enzim glial yang bertanggungjawab untuk sintesis asid quinolinic dalam striatum penyakit Huntington meningkat kira-kira 5 kali ganda berbanding normal, manakala aktiviti enzim yang memastikan degradasi asid quinolinic meningkat dalam penyakit Huntington sebanyak 20-50% sahaja. Oleh itu, sintesis asid quinolinic mungkin meningkat dalam penyakit Huntington.
Kajian reseptor asid amino pengujaan (EAA) dalam penyakit Huntington telah mendedahkan pengurangan ketara dalam bilangan reseptor glutamat NMDA, AMPA, kainat dan metabotropik dalam striatum, serta reseptor AMPA dan kainate dalam korteks serebrum. Pada peringkat akhir penyakit Huntington, reseptor NMDA hampir tidak hadir, manakala pada peringkat praklinikal dan awal, pengurangan ketara dalam bilangan reseptor ini telah dicatatkan.
Sensitiviti terpilih. Dalam penyakit Huntington, beberapa jenis sel striatal hilang secara selektif. Neuron berduri sederhana, yang menonjol ke segmen luar globus pallidus dan mengandungi GABA dan enkephalin, mati sangat awal dalam penyakit ini, begitu juga neuron yang mengandungi GABA dan bahan P dan mengunjur ke bahagian retikular substantia nigra. Kehilangan neuron yang mengandungi GABA dan enkephalin dan menonjol ke segmen luar globus pallidus menghalang struktur ini, yang seterusnya membawa kepada perencatan aktif nukleus subthalamic. Penurunan aktiviti nukleus subthalamic nampaknya boleh menjelaskan pergerakan koreiform yang berlaku dalam penyakit Huntington. Telah lama diketahui bahawa lesi fokus nukleus subthalamic boleh menyebabkan korea. Kehilangan neuron GABA dan bahan P yang menonjol ke substantia nigra pars reticularis berkemungkinan bertanggungjawab terhadap gangguan okulomotor yang dilihat dalam penyakit Huntington. Laluan ini biasanya menghalang neuron substantia nigra pars reticularis yang menonjol ke kolikulus superior, yang seterusnya mengawal selia saccades. Dalam penyakit Huntington juvana, laluan yang disebutkan di atas lebih teruk terjejas dan, sebagai tambahan, unjuran striatal ke segmen dalaman globus pallidus hilang lebih awal.
Protein huntingtin, yang dikodkan oleh gen yang mutasinya menyebabkan penyakit Huntington, ditemui dalam pelbagai struktur otak dan tisu lain. Huntingtin biasanya ditemui terutamanya dalam sitoplasma neuron. Protein ditemui dalam kebanyakan neuron di otak, tetapi data terkini menunjukkan bahawa kandungannya lebih tinggi dalam neuron matriks berbanding neuron striosomal, dan lebih tinggi dalam neuron unjuran daripada interneuron. Oleh itu, sensitiviti selektif neuron berkorelasi dengan kandungan huntingtin mereka, yang biasanya terdapat dalam populasi neuron tertentu.
Seperti dalam otak pesakit dengan penyakit Huntington, pada tikus transgenik untuk serpihan N-terminal gen penyakit Huntington dengan bilangan ulangan yang diperluaskan, huntingtin membentuk agregat padat dalam nukleus neuron. Kemasukan intranuklear ini terbentuk dalam neuron unjuran striatal (tetapi tidak dalam interneuron). Dalam tikus transgenik, kemasukan terbentuk beberapa minggu sebelum permulaan gejala. Data ini mencadangkan bahawa protein huntingtin yang mengandungi lebih banyak sisa glutamin yang kemasukannya mengekod ulangan trinukleotida, atau serpihannya, terkumpul di dalam nukleus dan akibatnya boleh menjejaskan kawalannya terhadap fungsi selular.
Gejala penyakit Huntington
Umur di mana gejala pertama muncul pada pesakit dengan penyakit Huntington adalah sukar untuk ditentukan dengan tepat, kerana penyakit itu menunjukkan dirinya secara beransur-ansur. Perubahan dalam personaliti dan tingkah laku, gangguan koordinasi ringan mungkin berlaku bertahun-tahun sebelum kemunculan gejala yang lebih jelas. Pada masa diagnosis ditubuhkan, kebanyakan pesakit mempunyai pergerakan koreik, koordinasi pergerakan halus yang terjejas, dan penjanaan kantung sukarela yang perlahan. Apabila penyakit itu berlanjutan, keupayaan untuk mengatur aktiviti seseorang terjejas, ingatan berkurangan, pertuturan menjadi sukar, gangguan okulomotor dan prestasi terjejas pergerakan terkoordinasi meningkat. Walaupun pada peringkat awal penyakit tidak ada perubahan dalam otot dan postur, kerana ia berkembang, postur dystonic mungkin berkembang, yang dari masa ke masa mungkin berubah menjadi gejala yang dominan. Pada peringkat akhir, pertuturan menjadi tidak jelas, menelan menjadi sangat sukar, berjalan menjadi mustahil. Penyakit Huntington biasanya berkembang selama 15-20 tahun. Pada peringkat terminal, pesakit tidak berdaya dan memerlukan penjagaan yang berterusan. Hasil maut tidak berkaitan secara langsung dengan penyakit utama, tetapi dengan komplikasinya, sebagai contoh, radang paru-paru.
Demensia dalam penyakit Huntington
Kod ICD-10
P02.2. Demensia dalam penyakit Huntington (G10).
Demensia berkembang sebagai salah satu manifestasi proses degeneratif-atropik sistemik dengan kerosakan utama pada sistem striatal otak dan nukleus subkoekal yang lain. Ia diwarisi secara autosomal dominan.
Sebagai peraturan, penyakit itu menampakkan diri pada dekad ketiga atau keempat kehidupan dengan hiperkinesis koreoform (terutamanya di muka, lengan, bahu, gaya berjalan), perubahan personaliti (jenis anomali personaliti yang teruja, histeris dan skizoid), gangguan psikotik (kemurungan khas dengan kesuraman, kemarahan, disforia; mood paranoid).
Kepentingan khusus untuk diagnostik ialah gabungan hiperkinesis koreoform, demensia dan beban keturunan. Berikut adalah khusus untuk demensia ini:
- perkembangan perlahan (purata 10-15 tahun): pemisahan antara baki keupayaan untuk menjaga diri sendiri dan ketidakcekapan intelek yang jelas dalam situasi yang memerlukan kerja mental yang produktif (pemikiran konsep, mempelajari perkara baru);
- menyatakan ketidaksamaan prestasi mental, yang berdasarkan gangguan perhatian yang teruk dan ketidakkonsistenan sikap pesakit ("berfikiran tersentak", serupa dengan hiperkinesis);
- atipikal pelanggaran jelas fungsi kortikal yang lebih tinggi;
- hubungan songsang antara peningkatan demensia dan keterukan gangguan psikotik.
Dengan mengambil kira bahagian psikotik yang tinggi (khayalan paranoid cemburu, penganiayaan) dan gangguan dysphoric dalam gambaran klinikal penyakit, rawatan dijalankan menggunakan pelbagai neuroleptik yang menyekat reseptor dopaminergik (derivatif fenotiazin dan butyrophenone) atau mengurangkan tahap dopamin dalam tisu (reserpine).
Haloperidol (2-20 mg / hari), tiapride (100-600 mg / hari) selama tidak lebih daripada tiga bulan, thioridazine (sehingga 100 mg / hari), reserpine (0.25-2 mg / hari), dan clonazepam anticonvulsant (1.5-6 mg / hari) digunakan. Ubat-ubatan ini membantu mengurangkan hiperkinesis, melancarkan ketegangan afektif, dan mengimbangi gangguan personaliti.
Rawatan pesakit dalam gangguan mental dijalankan dengan mengambil kira sindrom utama, umur dan keadaan umum pesakit. Dalam rawatan pesakit luar, prinsip terapi adalah sama (terapi penyelenggaraan berterusan gangguan pergerakan, perubahan ubat secara berkala). Dos neuroleptik yang lebih rendah digunakan dalam rawatan pesakit luar.
Langkah-langkah pemulihan untuk demensia ringan dan sederhana termasuk terapi pekerjaan, psikoterapi, dan latihan kognitif. Ia adalah perlu untuk bekerja dengan ahli keluarga dan memberikan sokongan psikologi kepada orang yang menjaga pesakit. Kaedah utama pencegahan penyakit adalah kaunseling perubatan dan genetik saudara terdekat pesakit dengan rujukan untuk analisis DNA apabila membuat keputusan untuk melahirkan anak.
Prognosis umumnya tidak menguntungkan. Perjalanan penyakit ini perlahan-lahan progresif, dan penyakit ini biasanya membawa kepada kematian selepas 10-15 tahun.
[ 18 ]
Apa yang mengganggumu?
Rawatan penyakit Huntington
Rawatan penyakit Huntington adalah simptomatik. Korea dan pergolakan boleh disekat sebahagiannya dengan neuroleptik (cth, chlorpromazine 25-300 mg secara lisan 3 kali sehari, haloperidol 5-45 mg secara lisan 2 kali sehari) atau reserpine 0.1 mg secara lisan sekali sehari. Dos dinaikkan kepada maksimum yang boleh diterima (sebelum kesan sampingan berlaku, seperti mengantuk, parkinsonisme; untuk reserpine, hipotensi). Matlamat terapi empirikal adalah untuk mengurangkan penghantaran glutamatergik melalui reseptor Nmethyl-O-aspartate dan mengekalkan pengeluaran tenaga dalam mitokondria. Rawatan yang bertujuan untuk meningkatkan GABA dalam otak adalah tidak berkesan.
Ujian genetik dan kaunseling adalah penting kerana gejala penyakit ini muncul selepas tahun-tahun melahirkan anak. Orang yang mempunyai sejarah keluarga yang positif dan mereka yang berminat dalam ujian dirujuk ke pusat khusus, dengan mengambil kira semua implikasi etika dan psikologi.
Rawatan simptomatik penyakit Huntington
Tiada rawatan berkesan yang boleh menghentikan perkembangan penyakit Huntington. Beberapa ujian pelbagai ubat telah dijalankan, tetapi tiada kesan ketara telah dicapai. Neuroleptik dan antagonis reseptor dopamin lain digunakan secara meluas untuk membetulkan gangguan mental dan pergerakan sukarela pada pesakit dengan penyakit Huntington. Pergerakan sukarela mencerminkan ketidakseimbangan antara sistem dopaminergik dan GABAergik. Oleh itu, neuroleptik digunakan untuk mengurangkan aktiviti dopaminergik yang berlebihan. Walau bagaimanapun, ubat-ubatan ini sendiri boleh menyebabkan kesan sampingan kognitif dan ekstrapiramidal yang ketara. Di samping itu, kecuali dalam kes di mana pesakit mengalami psikosis atau pergolakan, keberkesanannya belum terbukti. Neuroleptik sering menyebabkan atau memburukkan disfagia atau gangguan pergerakan lain. Neuroleptik generasi baru seperti risperidone, clozapine dan olanzapine mungkin amat berguna dalam rawatan penyakit Huntington kerana ia menyebabkan lebih sedikit kesan sampingan ekstrapiramidal tetapi boleh mengurangkan gejala paranoid atau meningkatkan kerengsaan.
Tetrabenazine dan reserpine juga mengurangkan aktiviti sistem dopaminergik dan boleh mengurangkan keterukan pergerakan sukarela pada peringkat awal penyakit. Walau bagaimanapun, ubat-ubatan ini boleh menyebabkan kemurungan. Oleh kerana penyakit itu sendiri sering menyebabkan kemurungan, kesan sampingan ini mengehadkan penggunaan reserpine dan tetrabenazine dengan ketara. Pada peringkat akhir penyakit, sel yang mengandungi reseptor dopamin mati, jadi keberkesanan antagonis reseptor dopamin menjadi lemah atau hilang.
Neuroleptik, antidepresan, dan anxiolytics digunakan untuk merawat psikosis, kemurungan, dan kerengsaan pada pesakit dengan penyakit Huntington, tetapi ia hanya perlu ditetapkan selagi pesakit benar-benar mengalami gejala ini. Ubat-ubatan yang mungkin membantu pada satu peringkat penyakit mungkin menjadi tidak berkesan atau bahkan berbahaya apabila penyakit itu berkembang.
Agonis reseptor GABA telah diuji pada pesakit dengan penyakit Huntington, memandangkan penyakit Huntington telah terbukti mempunyai penurunan ketara dalam tahap GABA dalam striatum, serta hipersensitiviti reseptor GABA di kawasan unjurannya. Benzodiazepin telah terbukti berkesan dalam kes-kes di mana pergerakan sukarela dan gangguan kognitif diburukkan lagi oleh tekanan dan kebimbangan. Dos rendah ubat-ubatan ini harus ditetapkan untuk mengelakkan kesan sedatif yang tidak diingini. Dalam kebanyakan pesakit dengan penyakit Huntington, tiada ubat yang membawa kepada peningkatan yang ketara dalam kualiti hidup.
Dalam penyakit Huntington yang awal dengan gejala parkinson, agen dopaminergik boleh dicuba, tetapi keberkesanannya terhad. Lebih-lebih lagi, levodopa boleh menyebabkan atau meningkatkan myoclonus pada pesakit ini. Pada masa yang sama, baclofen boleh mengurangkan ketegaran pada sesetengah pesakit dengan penyakit Huntington.
[ 26 ], [ 27 ], [ 28 ], [ 29 ]
Rawatan pencegahan (neuroprotektif) penyakit Huntington
Walaupun kecacatan genetik dalam penyakit Huntington diketahui, bagaimana ia membawa kepada degenerasi neuron terpilih masih tidak jelas. Adalah dihipotesiskan bahawa terapi pencegahan yang bertujuan untuk mengurangkan tekanan oksidatif dan excitotoxicity mungkin berpotensi memperlahankan atau menghentikan perkembangan penyakit. Keadaan ini mungkin agak serupa dengan degenerasi hepatolentikular, di mana kecacatan genetik kekal tidak diketahui selama bertahun-tahun, tetapi terapi pencegahan yang bertujuan untuk kesan sekunder, pengumpulan tembaga, telah "sembuh." Dalam hal ini, hipotesis bahawa penyakit Huntington dikaitkan dengan gangguan metabolisme tenaga dan kematian sel akibat excitotoxicity telah menarik perhatian khusus. Penyakit itu sendiri boleh menyebabkan kematian sel akibat pengagregatan intranuklear serpihan terminal N huntingtin, yang mengganggu fungsi selular dan metabolik. Proses ini mungkin menjejaskan beberapa kumpulan neuron pada tahap yang lebih besar daripada yang lain disebabkan oleh kepekaan yang lebih tinggi terhadap kerosakan eksitotoksik. Dalam kes ini, terapi pencegahan dengan antagonis reseptor asid amino rangsang atau agen yang menghalang kerosakan radikal bebas akan dapat mencegah atau melambatkan permulaan dan perkembangan penyakit. Dalam model makmal amyotrophic lateral sclerosis, telah ditunjukkan bahawa agen antioksidan dan antagonis reseptor (RAA) mampu melambatkan perkembangan penyakit. Pendekatan yang sama mungkin berkesan dalam penyakit Huntington. Percubaan klinikal antagonis reseptor glutamat dan agen yang meningkatkan fungsi kompleks II rantai pengangkutan elektron mitokondria sedang dijalankan.
[ 30 ], [ 31 ], [ 32 ], [ 33 ], [ 34 ], [ 35 ], [ 36 ], [ 37 ], [ 38 ], [ 39 ]