Pakar perubatan artikel itu

Penerbitan baru
Sindrom Usher
Ulasan terakhir: 04.07.2025

Semua kandungan iLive disemak secara perubatan atau fakta diperiksa untuk memastikan ketepatan faktual sebanyak mungkin.
Kami mempunyai garis panduan sumber yang ketat dan hanya memautkan ke tapak media yang bereputasi, institusi penyelidikan akademik dan, apabila mungkin, dikaji semula kajian secara medis. Perhatikan bahawa nombor dalam kurungan ([1], [2], dan lain-lain) boleh diklik pautan ke kajian ini.
Jika anda merasakan bahawa mana-mana kandungan kami tidak tepat, ketinggalan zaman, atau tidak dipersoalkan, sila pilih dan tekan Ctrl + Enter.
Sindrom Usher adalah penyakit keturunan yang menunjukkan dirinya sebagai pekak lengkap sejak lahir, serta buta progresif dengan usia. Kehilangan penglihatan dikaitkan dengan retinitis pigmentosa, proses degenerasi pigmen retina. Ramai penghidap sindrom Usher juga mengalami masalah keseimbangan yang teruk.
Epidemiologi
Terima kasih kepada penyelidikan, adalah mungkin untuk menentukan bahawa sindrom Usher menjejaskan kira-kira 8% daripada kanak-kanak pekak-bisu yang diperiksa (ujian dijalankan di institusi khas untuk orang pekak-bisu). Retinitis pigmen diperhatikan dalam 6-10% pesakit yang menderita pekak kongenital, yang seterusnya, diperhatikan pada kira-kira 30% orang dengan penyakit retina pigmen.
Adalah dipercayai bahawa penyakit ini menunjukkan dirinya dalam kira-kira 3-10 orang daripada 100 ribu di seluruh dunia. Ia boleh diperhatikan pada wanita dan lelaki secara sama rata. Kira-kira 5-6% penduduk dunia mengalami sindrom ini. Kira-kira 10% daripada semua kes pekak mendalam kanak-kanak berlaku disebabkan oleh sindrom Usher I, serta jenis II.
Di Amerika Syarikat, jenis 1 dan 2 adalah jenis yang paling biasa. Bersama-sama, mereka menyumbang kira-kira 90 hingga 95 peratus daripada semua kes sindrom Usher pada kanak-kanak.
Punca Sindrom Usher
Sindrom Usher jenis I, II, dan III mempunyai punca resesif autosomal, manakala jenis IV dianggap sebagai gangguan kromosom X. Punca-punca buta dan pekak yang berlaku dengan sindrom ini masih belum cukup dikaji. Diandaikan bahawa penghidap penyakit ini hipersensitif kepada komponen yang boleh merosakkan struktur DNA. Di samping itu, penyakit ini mungkin dikaitkan dengan gangguan sistem imun, tetapi dalam kes ini, tidak ada gambaran yang tepat tentang proses ini.
Pada tahun 1989, keabnormalan kromosom pertama kali dikenal pasti pada pesakit dengan penyakit jenis II, yang mungkin pada masa hadapan membawa kepada cara untuk mengasingkan gen yang menyebabkan sindrom. Ia juga mungkin untuk mengenal pasti gen ini dalam pembawa dan membangunkan ujian genetik pranatal khas.
 [ 8 ]
[ 8 ]
Faktor-faktor risiko
Sindrom ini diwarisi apabila kedua-dua ibu bapa terjejas, iaitu, ia diwarisi oleh jenis resesif. Seorang kanak-kanak juga boleh mewarisi penyakit ini jika ibu bapanya adalah pembawa gen. Jika kedua-dua ibu bapa masa depan mempunyai gen ini, maka kebarangkalian untuk mempunyai bayi dengan sindrom ini adalah 1 dalam 4. Seseorang yang mempunyai hanya satu gen untuk sindrom itu dianggap pembawa, tetapi tidak mempunyai gejala gangguan. Pada masa kini, masih belum dapat ditentukan sama ada seseorang mempunyai gen untuk penyakit ini.
Sekiranya seorang kanak-kanak dilahirkan kepada ibu bapa, salah seorang daripadanya tidak mempunyai gen sedemikian, maka kebarangkalian bahawa dia akan mewarisi sindrom itu sangat rendah, tetapi dia pasti akan menjadi pembawa.
Gejala Sindrom Usher
Gejala sindrom Usher termasuk kehilangan pendengaran dan pengumpulan abnormal sel berpigmen dalam struktur mata. Pesakit kemudiannya mengalami degenerasi retina, yang menyebabkan kemerosotan penglihatan dan akhirnya kehilangan penglihatan dalam kes yang paling teruk.
Kehilangan pendengaran sensorineural boleh menjadi ringan atau lengkap dan biasanya tidak berkembang sejak lahir. Walau bagaimanapun, penyakit pigmen retina boleh mula berkembang pada zaman kanak-kanak atau kemudian. Keputusan ujian telah menunjukkan bahawa ketajaman penglihatan pusat boleh dikekalkan selama bertahun-tahun, walaupun apabila penglihatan periferi merosot (keadaan yang dipanggil "penglihatan terowong").
Ini adalah manifestasi utama penyakit ini, yang kadang-kadang boleh ditambah dengan gangguan lain, seperti psikosis dan gangguan mental lain, masalah dengan telinga dalam dan/atau katarak.
Borang
Semasa penyelidikan, 3 jenis penyakit ini dikenal pasti, serta bentuk ke-4, yang agak jarang berlaku.
Jenis I penyakit ini dicirikan oleh pekak lengkap kongenital, serta gangguan keseimbangan. Selalunya, kanak-kanak seperti itu mula berjalan hanya pada usia 1.5 tahun. Kemerosotan penglihatan biasanya bermula pada usia 10 tahun, dan perkembangan akhir keadaan rabun malam bermula pada usia 20 tahun. Kanak-kanak dengan jenis penyakit ini mungkin mengalami kemerosotan progresif penglihatan periferi.
Dalam penyakit jenis II, pekak sederhana atau kongenital diperhatikan. Dalam kes ini, kemerosotan dalam pekak separa selalunya tidak berlaku lagi. Retinitis pigmen mula berkembang sekitar akhir remaja atau selepas 20 tahun. Perkembangan rabun malam biasanya bermula pada 29-31 tahun. Kemerosotan ketajaman penglihatan dalam kes patologi jenis II umumnya berkembang sedikit lebih perlahan daripada jenis I.
Jenis III penyakit ini dicirikan oleh kehilangan pendengaran yang progresif, biasanya bermula semasa akil baligh, serta perkembangan beransur-ansur dalam tempoh yang sama (sedikit lewat daripada kehilangan pendengaran) retinitis pigmentosa, yang boleh menjadi faktor dalam perkembangan buta progresif.
Manifestasi patologi jenis IV terutamanya berlaku pada lelaki. Dalam kes ini, gangguan progresif dan kehilangan pendengaran dan penglihatan juga diperhatikan. Bentuk ini sangat jarang berlaku dan biasanya mempunyai sifat kromosom X.
Diagnostik Sindrom Usher
Diagnosis sindrom Usher dibuat berdasarkan gabungan pesakit yang diperhatikan iaitu pekak secara tiba-tiba dan kehilangan penglihatan yang progresif.
Ujian
Ujian genetik khas boleh diarahkan untuk mengesan mutasi.
Sebelas lokus genetik telah ditemui yang boleh menyebabkan perkembangan sindrom Usher, dan sembilan gen telah dikenal pasti yang pasti punca gangguan:
- Jenis 1: MY07A, USH1C, Cdh23, Pcdh15, SANS.
- Jenis 2: ush2a, VLGR1, WHRN.
- Sindrom Usher jenis 3: USH3A.
Para saintis NIDCD, bersama-sama dengan rakan sekerja dari universiti di New York dan Israel, telah mengenal pasti mutasi yang dipanggil R245X dalam gen Pcdh15 yang menyumbang peratusan besar sindrom Usher jenis 1 dalam populasi Yahudi.
Untuk mengetahui tentang makmal yang menjalankan ujian klinikal, lawati https://www.genetests.org dan cari direktori makmal untuk "sindrom Usher."
Untuk mengetahui tentang ujian klinikal sedia ada yang termasuk ujian genetik untuk sindrom Usher, lawati https://www.clinicaltrials.gov dan cari "Sindrom Usher" atau "ujian genetik sindrom Usher."
[ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ], [ 30 ]
Diagnostik instrumental
Terdapat beberapa kaedah diagnostik instrumental:
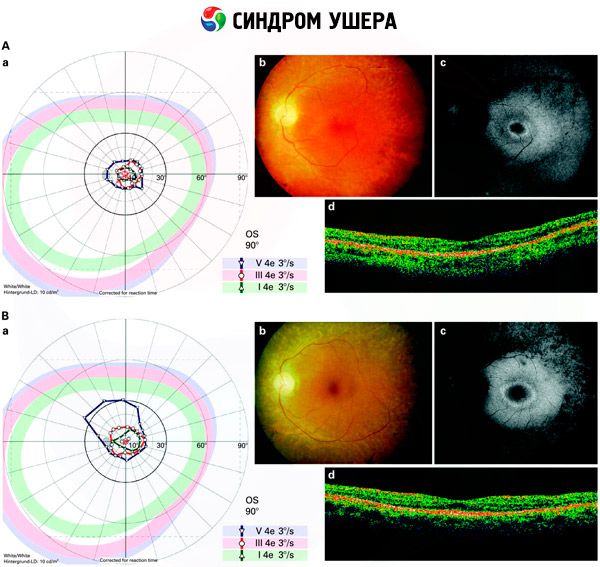
- Pemeriksaan fundus untuk mengesan kehadiran bintik-bintik pigmen pada retina, serta penyempitan saluran retina;
- Electroretinogram, yang membolehkan untuk mengesan penyimpangan degeneratif awal dalam retina mata. Ia menunjukkan kepupusan laluan elektroradiografi;
- Elektronystagmogram (ENG) mengukur pergerakan mata yang tidak disengajakan yang boleh menunjukkan kehadiran ketidakseimbangan.
- Audiometri, yang digunakan untuk menentukan kehadiran pekak dan keparahannya.
Diagnosis pembezaan
Sindrom Usher mesti dibezakan daripada beberapa gangguan yang serupa.
Sindrom Hallgren, yang dicirikan oleh kehilangan pendengaran kongenital dan kehilangan penglihatan progresif (katarak dan nystagmus juga berkembang). Gejala tambahan termasuk ataxia, gangguan psikomotor, psikosis, dan terencat akal.
Sindrom Alstrom, iaitu penyakit keturunan di mana retina merosot, mengakibatkan kehilangan penglihatan pusat. Sindrom ini dikaitkan dengan obesiti kanak-kanak. Pada masa yang sama, diabetes mellitus dan kehilangan pendengaran mula berkembang selepas 10 tahun.
Rubella pada wanita hamil pada trimester pertama boleh menyebabkan pelbagai kelainan dalam perkembangan kanak-kanak. Antara akibat daripada kelainan tersebut adalah kehilangan pendengaran, serta (atau) masalah dengan penglihatan, dan sebagai tambahan kepada ini, pelbagai kecacatan perkembangan.
Siapa yang hendak dihubungi?
Rawatan Sindrom Usher
Pada masa ini tiada ubat untuk sindrom Usher. Oleh itu, terapi dalam kes ini terutamanya terdiri daripada melambatkan proses kehilangan penglihatan, serta mengimbangi kehilangan pendengaran. Kaedah rawatan yang mungkin termasuk:
- Mengambil vitamin A (sesetengah pakar oftalmologi percaya bahawa dos tinggi vitamin A palmitat boleh melambatkan, tetapi tidak menghentikan, perkembangan retinitis pigmentosa);
- Implantasi peranti elektronik khas ke dalam telinga pesakit (alat bantu pendengaran, implan koklea.
Pakar oftalmologi mengesyorkan bahawa kebanyakan orang dewasa dengan bentuk biasa retinitis pigmentosa mengambil 15,000 IU (unit antarabangsa) vitamin A palmitat setiap hari di bawah pengawasan. Oleh kerana orang dengan sindrom Usher jenis 1 tidak termasuk dalam kajian, dos vitamin A yang tinggi tidak disyorkan untuk kumpulan pesakit ini. Orang yang sedang mempertimbangkan untuk mengambil vitamin A harus membincangkan pilihan rawatan ini dengan doktor mereka. Cadangan lain untuk pilihan rawatan ini termasuk:
- Mengubah diet anda untuk memasukkan makanan yang tinggi vitamin A.
- Wanita yang merancang untuk hamil harus berhenti mengambil dos vitamin A yang tinggi tiga bulan sebelum mereka merancang untuk hamil kerana peningkatan risiko kecacatan kelahiran.
- Wanita yang hamil harus berhenti mengambil dos vitamin A yang tinggi kerana peningkatan risiko kecacatan kelahiran.
Ia juga penting untuk menyesuaikan kanak-kanak sedemikian dengan kehidupan sosial. Ini memerlukan bantuan guru pendidikan khas dan ahli psikologi. Sekiranya pesakit telah mula mengalami kehilangan penglihatan yang progresif, dia harus diajar menggunakan bahasa isyarat.
Ramalan
Sindrom Usher mempunyai prognosis yang tidak baik. Bidang visual dan ketajamannya mula merosot dalam tempoh 20-30 tahun pada kebanyakan pesakit dengan penyakit ini dari sebarang jenis. Dalam sesetengah kes, kehilangan penglihatan dua hala sepenuhnya berlaku. Kehilangan pendengaran, yang selalu disertai dengan kebisuan, berkembang dengan cepat untuk menyelesaikan kehilangan pendengaran dua hala.