Pakar perubatan artikel itu

Penerbitan baru
Alkaptonuria adalah kelainan enzim kongenital
Ulasan terakhir: 04.07.2025

Semua kandungan iLive disemak secara perubatan atau fakta diperiksa untuk memastikan ketepatan faktual sebanyak mungkin.
Kami mempunyai garis panduan sumber yang ketat dan hanya memautkan ke tapak media yang bereputasi, institusi penyelidikan akademik dan, apabila mungkin, dikaji semula kajian secara medis. Perhatikan bahawa nombor dalam kurungan ([1], [2], dan lain-lain) boleh diklik pautan ke kajian ini.
Jika anda merasakan bahawa mana-mana kandungan kami tidak tepat, ketinggalan zaman, atau tidak dipersoalkan, sila pilih dan tekan Ctrl + Enter.
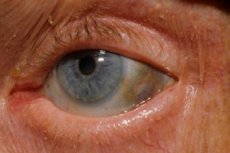
Salah satu gangguan metabolik yang sangat jarang berlaku, alkaptonuria, merujuk kepada anomali kongenital dalam metabolisme asid amino tirosin.
Sindrom ini juga boleh dipanggil kekurangan homogentisate oxidase, homogentisinuria, ochronosis keturunan, atau penyakit kencing hitam.[ 1 ]
Epidemiologi
Menurut statistik, terdapat tidak lebih daripada sembilan kes alkaptonuria bagi setiap 1 juta orang. Dan di kebanyakan negara Eropah, terdapat satu kes setiap 100-250 ribu kelahiran hidup.
Di kalangan negara Eropah, pengecualian adalah Slovakia (terutamanya wilayah barat laut yang agak kecil), di mana kelaziman alkaptonuria adalah satu kes bagi setiap 19,000 bayi baru lahir. Ini berkemungkinan besar disebabkan oleh fakta bahawa di kalangan keluarga Roma Slovakia yang tinggal di sana, tahap pembiakan dalam (perkahwinan antara sepupu) adalah yang tertinggi di Eropah: 10-14%. [ 2 ]
Punca alkaptonuria
Punca sebenar alkaptonuria, sebagai gangguan kongenital katabolisme (pecahan metabolik) tirosin asid α-amino aromatik (homocyclic), telah ditubuhkan: jenis gangguan metabolik ini adalah akibat daripada mutasi heterozigot homozigot atau kompaun salah satu daripada beribu-ribu gen pada kromosom 3, lebih tepat lagi pada gen HG3q2, lebih tepat lagi. lengan kromosom. Gen ini mengekod jujukan nukleotida enzim hati homogentisate-1,2-dioxygenase [ 3 ] (juga dipanggil homogentisic acid oxidase atau homogentisate oxidase) - metalloprotein yang mengandungi besi yang diperlukan untuk salah satu peringkat pemecahan tirosin dalam badan. [ 4 ], [ 5 ]
Oleh itu, alkaptonuria adalah kecacatan enzim homogentisate-1,2-dioxygenase, atau lebih tepat lagi, hasil daripada kekurangan yang ditentukan secara genetik atau ketiadaan sepenuhnya. [ 6 ]
Sebagai kekurangan enzim kongenital, alkaptonuria diwarisi sebagai sifat resesif autosomal, iaitu, untuk alkaptonuria berlaku pada kanak-kanak, kedua-dua ibu bapa mesti mempunyai gen yang diubah suai untuk enzim itu, kerana setiap daripada mereka mewariskan kepada kanak-kanak hanya satu salinan gen daripada dua yang tersedia.
Menurut data terkini, terdapat lebih daripada dua ratus varian pengubahsuaian gen HGD, dan mutasi missense, translokasi dan splicing paling kerap diperhatikan.
Faktor-faktor risiko
Satu-satunya faktor risiko untuk membangunkan enzimopati kongenital ini ialah kehadirannya dalam sejarah keluarga dan pewarisan dua salinan gen HGD yang diubah suai, jika ibu bapa tidak menunjukkan alkaptonuria (risiko penghantaran anomali ialah 25%), atau salah seorang daripada ibu bapa mengalami gangguan ini. [ 7 ]
Patogenesis
Tyrosine memainkan peranan penting dalam sintesis protein, penghasilan kromoprotein - pigmen kulit melanin, serta hormon tiroid dan neurotransmitter katekolamin.
Mekanisme untuk mengawal jumlah tirosin dalam sel adalah sangat kompleks, dan badan menormalkan kandungan berlebihannya dengan memecahkannya. Proses katabolisme tirosin, seperti semua asid amino aromatik, adalah berbilang peringkat dan berlaku dalam beberapa peringkat. Setiap peringkat pemecahan metabolik tirosin berlaku dengan penyertaan enzim tertentu dan pembentukan sebatian perantaraan.
Jadi, pertama asid amino dipecahkan kepada para-hydroxyphenylpyruvate, yang ditukar kepada alkaptone - 2,5-dihydroxyphenylacetic atau asid homogentisic. Kemudian alkaptone harus diubah menjadi asid maleasetik, tetapi ini tidak berlaku. [ 8 ]
Dan patogenesis alkaptonuria terdiri daripada pemberhentian tindak balas biokimia katabolisme tirosin pada peringkat pembentukan asid homogentisic: tidak ada enzim yang diperlukan untuk memecahkannya - homogentisate oxidase.
Asid homogentisic tidak digunakan oleh badan dan boleh terkumpul dengan perkumuhan melalui buah pinggang. Di samping itu, ia dioksidakan kepada benzoquinoacetate (asid benzoquinoneacetic), yang, dengan mengikat molekul tisu dan cecair badan, membentuk sebatian biopolimer berwarna seperti melanin.
Pengumpulan produk perantaraan ini dalam tisu membawa kepada gangguan struktur kolagen tisu tulang rawan, yang mengurangkan keanjalannya - dengan kemunculan banyak tanda klinikal alkaptonuria dan perkembangan komplikasi.
Gejala alkaptonuria
Alkaptonuria pada bayi baru lahir dan bayi dicirikan oleh kegelapan air kencing. Apabila terdedah kepada udara, air kencing pada lampin, lampin, dan seluar dalam menjadi coklat gelap; ini disebabkan oleh pengumpulan dan pembebasan asid homogentisic, yang teroksida kepada benzoquinoacetate. [ 9 ]
Sekiranya tiada gejala lain, alkaptonuria pada kanak-kanak kecil sering tidak dikenali tepat pada masanya, kerana air kencing mungkin menjadi gelap selepas beberapa jam buang air kecil. Menurut beberapa data, hanya satu perlima kanak-kanak di bawah 12 bulan yang dilahirkan dengan kekurangan enzim ini dikenal pasti dalam tetapan klinikal. Oleh itu, amat penting bagi ibu bapa untuk memberi perhatian terhadap penjagaan bayi mereka.
Selain itu, tanda-tanda awal termasuk pigmentasi (warna kelabu kebiruan) pada sklera mata dan rawan telinga dan hidung, yang sering dipanggil ochronosis.[ 10 ]
Dari masa ke masa, gejala lain muncul:
- pigmentasi kulit yang teruk pada tulang pipi, ketiak dan alat kelamin;
- mengotorkan pakaian apabila bersentuhan dengan bahagian badan yang berpeluh;
- serangan kelemahan umum;
- suara serak.
Perlu diingat bahawa alkaptonuria dan ochronosis, seperti yang dinyatakan di atas, adalah nama yang sinonim untuk gangguan katabolisme tirosin yang sama.
Penyakit kencing sirap maple dan alkaptonuria. Penyakit kencing sirap maple kongenital atau leucinosis juga merupakan gangguan metabolik, mempunyai corak warisan yang sama, malah mutasi berlaku pada kromosom yang sama, tetapi menjejaskan gen pengekodan kompleks enzim rantai bercabang α-keto dehidrogenase asid. Disebabkan ini, badan tidak boleh memecahkan komponen tertentu protein, khususnya, asid amino leucine, isoleucine dan valine. Dengan penyakit ini, air kencing (dan tahi telinga) mempunyai bau yang manis; di samping itu, gambaran klinikal jenis asidemia organik ini termasuk hipopigmentasi, turun naik dalam tekanan darah, sawan, muntah dan cirit-birit, penurunan paras glukosa darah, ketoasidosis, halusinasi, dll. Kadar kematian pada kanak-kanak agak tinggi; pada orang dewasa, tanpa rawatan, koma dan kematian mungkin berlaku akibat edema serebrum.
Albinisme dan alkaptonuria "disatukan" hanya oleh tirosin. Albinisme, termasuk oculocutaneous, disebabkan oleh mutasi genetik yang menjejaskan pengeluaran pigmen melanin. Perubahan kongenital dicatatkan dalam gen TYR pada kromosom 11 (11q14.3), yang mengkodkan tyrosinase, enzim melanosom yang mengandungi tembaga yang diperlukan untuk pembentukan pigmen kulit berdasarkan produk metabolisme tirosin. Penyakit ini lebih biasa daripada alkaptonuria.
Komplikasi dan akibatnya
Disebabkan oleh tindakan metabolit perantaraan tyrosine - asid homogentisic dan benzoquinoneacetic - akibat dan komplikasi alkaptonuria muncul disebabkan oleh pemendapan polimer berpigmen reaktif, pemusnahan fibril kolagen dan kemerosotan keadaan rawan (dengan penurunan ketahanan mereka terhadap tekanan mekanikal).
Selama bertahun-tahun, pada masa dewasa, arthritis degeneratif dan osteoarthritis sendi besar (pinggul, sacroiliac dan lutut) berkembang; ruang intervertebral sempit (terutamanya di tulang belakang lumbar dan toraks) - dengan kalsifikasi dan pembentukan osteofit; ketumpatan tisu plat tulang subkondral berkurangan, dan tulang yang mendasari mungkin mengalami pembentukan semula patologi dengan pembentukan pertumbuhan dan ubah bentuk. [ 11 ]
Kerosakan pada injap jantung (aorta dan mitral) dan arteri koronari boleh diperhatikan - dengan tanda-tanda penyakit jantung koronari, serta pembentukan batu di buah pinggang dan kelenjar prostat - disebabkan oleh kalsifikasi yang sama. [ 12 ], [ 13 ]
Diagnostik alkaptonuria
Biasanya, diagnosis gangguan metabolik kongenital adalah berdasarkan kajian cecair biologi badan.
Berdasarkan ujian dan tindak balas apakah alkaptonuria boleh didiagnosis? Ujian air kencing diperlukan untuk mengesan asid homogentisic dan menentukan tahapnya (normal – 20-30 mg sehari, dinaikkan – 3-8 g). Sampel air kencing diperiksa dengan kromatografi gas atau spektrometri jisim, menggunakan kromatografi cecair; ujian saringan untuk kehadiran besi klorida dalam air kencing adalah mungkin. [ 14 ]
Terdapat juga kaedah untuk diagnostik pantas - menentukan alkapton dalam kotoran air kencing kering di atas kertas (mengikut keamatan warna).
Apabila menjelaskan diagnosis, diagnostik instrumental (radiografi) melibatkan mengenal pasti tanda radiologi osteoarthritis dan patologi sendi lain pada pesakit.
Diagnosis disahkan oleh kaedah genetik molekul untuk mendiagnosis penyakit keturunan, seperti ujian genetik dan penjujukan DNA. [ 15 ]
Diagnosis pembezaan
Diagnosis pembezaan termasuk hemochromatosis dan kegagalan hati akut bayi baru lahir, melaninuria, porfiria terputus-putus akut, limfohistiositosis hemophagocytic, patologi mitokondria primer, arthritis rheumatoid, ankylosing spondylitis.
Siapa yang hendak dihubungi?
Rawatan alkaptonuria
Rawatan utama untuk alkaptonuria ialah pemberian oral dos besar (sekurang-kurangnya 1000 mg sehari) asid askorbik. Pada kanak-kanak, ini meningkatkan perkumuhan asid homogentisic dalam air kencing, dan pada orang dewasa, ia mengurangkan kandungan urin derivatifnya, asid benzoquinoneacetic, dan melambatkan pengikatannya pada struktur tisu penghubung sendi dan kolagen. [ 16 ]
Klinik Eropah Barat sedang menguji ubat Nitisinone (Orfalin), ubat daripada kumpulan metabolit yang menghalang peringkat kedua katabolisme tirosin: transformasi para-hydroxyphenylpyruvate menjadi asid homogentisic. Walau bagaimanapun, penggunaan agen farmakologi ini membawa kepada pengumpulan tirosin dan boleh menyebabkan kesan sampingan yang teruk, termasuk kelegapan kornea dan fotofobia, pendarahan hidung dan perut, kegagalan hati, perubahan dalam darah, dll. Namun begitu, di Amerika Syarikat, Nitisinone diluluskan oleh FDA untuk rawatan tyrosinemia jenis I. [17 ], [ 18 ]
Oleh itu, rawatan fisioterapi - terapi senaman untuk meningkatkan kekuatan otot dan meningkatkan mobiliti sendi, terapi balneo dan peloid untuk mengehadkan kesakitan - dijalankan untuk masalah sendi yang disebabkan oleh alkaptonuria.
Walaupun tirosin bukan sahaja dibekalkan oleh makanan tetapi juga dihasilkan dalam badan, pesakit dengan alkaptonuria disyorkan untuk mengikuti diet rendah protein dan mengehadkan penggunaan makanan yang kaya dengan tirosin, terutamanya daging lembu dan daging babi, produk tenusu (terutamanya keju), kekacang, kacang dan biji.
Pencegahan
Pencegahan mutasi gen adalah mustahil, tetapi untuk mengelakkan kelahiran kanak-kanak dengan risiko tinggi gangguan kongenital, terdapat kaunseling genetik perubatan, yang diperlukan sebelum kehamilan yang dirancang untuk pasangan yang mempunyai sejarah keluarga termasuk penyakit keturunan. [ 19 ]
Ramalan
Hasil maut daripada alkaptonuria sangat jarang berlaku, dan kematian mungkin disebabkan oleh komplikasi serius yang melibatkan jantung dan buah pinggang. Jadi jangka hayat keseluruhan penghidap alkaptonuria adalah baik.
Tetapi kualiti hidup berkurangan kerana kesakitan yang teruk pada sendi atau tulang belakang dengan batasan mobiliti yang ketara, selalunya progresif.