Pakar perubatan artikel itu

Penerbitan baru
Penyakit Charcot-Marie-Tooth.
Ulasan terakhir: 12.07.2025

Semua kandungan iLive disemak secara perubatan atau fakta diperiksa untuk memastikan ketepatan faktual sebanyak mungkin.
Kami mempunyai garis panduan sumber yang ketat dan hanya memautkan ke tapak media yang bereputasi, institusi penyelidikan akademik dan, apabila mungkin, dikaji semula kajian secara medis. Perhatikan bahawa nombor dalam kurungan ([1], [2], dan lain-lain) boleh diklik pautan ke kajian ini.
Jika anda merasakan bahawa mana-mana kandungan kami tidak tepat, ketinggalan zaman, atau tidak dipersoalkan, sila pilih dan tekan Ctrl + Enter.
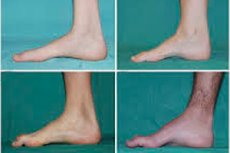
Atrofi otot peroneal, sindrom atau penyakit Charcot-Marie-Tooth ialah sekumpulan penyakit keturunan kronik dengan kerosakan pada saraf periferi.
Menurut ICD-10, dalam bahagian penyakit sistem saraf, kod untuk penyakit ini ialah G60.0 (motor keturunan dan neuropati deria). Ia juga termasuk dalam senarai penyakit anak yatim.
Epidemiologi
Menurut statistik klinikal, kelaziman semua jenis penyakit Charcot-Marie-Tooth bagi setiap 100 ribu penduduk adalah 19 kes (mengikut sumber lain, satu kes setiap 2.5-10 ribu penduduk).
Jenis CMT 1 menyumbang kira-kira dua pertiga daripada kes (satu kes setiap 5,000 hingga 7,000 populasi), dan hampir 70% daripadanya dikaitkan dengan pertindihan gen PMP22. Lebih daripada 1.2 juta orang di seluruh dunia menghidap jenis penyakit ini.
Insiden CMT jenis 4 dianggarkan pada 1-5 kes bagi setiap 10,000 kanak-kanak. [ 1 ]
Punca Penyakit Charcot-Marie-Tooth
Mengikut klasifikasi sindrom polyneuropathic, atrofi otot peroneal (fibular), amyotrophy neural Charcot-Marie-Tooth atau penyakit Charcot-Marie-Tooth (disingkat CMT) merujuk kepada polineuropati motor-deria yang ditentukan secara genetik. [ 2 ]
Iaitu, punca kejadiannya adalah mutasi genetik. Dan bergantung kepada sifat penyimpangan genetik, jenis atau jenis utama sindrom ini dibezakan: demielinasi dan axonal. Kumpulan pertama termasuk penyakit Charcot-Marie-Tooth jenis 1 (CMT1), yang berlaku disebabkan oleh pertindihan gen PMP22 pada kromosom 17, yang mengkodkan protein myelin periferal transmembran 22. Akibatnya, demielinasi segmental sarung akson (proses sel saraf) dan penurunan dalam kelajuan pengaliran isyarat saraf. Di samping itu, mutasi mungkin dalam beberapa gen lain.
Bentuk akson ialah penyakit Charcot-Marie-Tooth jenis 2 (CMT2), yang mempengaruhi akson itu sendiri dan dikaitkan dengan perubahan patologi dalam gen MFN2 di lokus 1p36.22, mengekod protein membran mitofusin-2, yang diperlukan untuk gabungan mitokondria dan pembentukan rangkaian mitokondria berfungsi di dalam sel saraf periferal. Terdapat lebih daripada sedozen subtipe CMT2 (dengan mutasi dalam gen tertentu).
Perlu diingatkan bahawa pada masa ini lebih daripada seratus gen telah dikenal pasti yang kerosakannya, dihantar melalui warisan, menyebabkan pelbagai subtipe penyakit Charcot-Marie-Tooth. Sebagai contoh, mutasi dalam gen RAB7 membawa kepada jenis CMT 2B; perubahan gen SH3TC2 (pengekodan salah satu protein membran sel Schwann) menyebabkan CMT jenis 4C, yang menunjukkan dirinya pada zaman kanak-kanak dan dicirikan oleh demielinasi neuron motor dan deria (terdapat sedozen setengah bentuk jenis 4 penyakit ini).
Jenis 3 CMT yang jarang berlaku (dipanggil sindrom Dejerine-Sottas) mula berkembang pada zaman kanak-kanak awal dan disebabkan oleh mutasi pada gen PMP22, MPZ, EGR2 dan lain-lain.
Apabila CMT jenis 5 berlaku pada usia 5-12 tahun, bukan sahaja neuropati motor (dalam bentuk paraparesis spastik pada bahagian bawah kaki) diperhatikan, tetapi juga kerosakan pada saraf optik dan pendengaran.
Kelemahan otot dan atrofi optik (dengan kehilangan penglihatan) serta masalah keseimbangan adalah tipikal bagi jenis CMT 6. Dan dalam penyakit Charcot-Marie-Tooth jenis 7 bukan sahaja neuropati motor-deria, tetapi juga penyakit retina dalam bentuk retinitis pigmentosa.
Lebih biasa di kalangan lelaki, penyakit CMT berkait-X atau Charcot-Marie-Tooth dengan tetraparesis anggota badan (kelemahan pergerakan kedua-dua lengan dan kaki) ialah jenis penyahmielinan dan dianggap berpunca daripada mutasi gen GJB1 pada lengan panjang kromosom X, yang mengekodkan sel penghubung 32 dan protein oligosmembrane, Schadendrondromembrane, yang mengkodekan sel penghubung 32, oligosmembrane. penghantaran isyarat saraf. [ 3 ]
Faktor-faktor risiko
Faktor risiko utama untuk CMT adalah sejarah keluarga penyakit ini, iaitu, dalam saudara terdekat.
Menurut pakar genetik, jika kedua-dua ibu bapa adalah pembawa gen resesif autosomal untuk penyakit Charcot-Marie-Tooth, risiko mempunyai anak yang akan menghidap penyakit ini ialah 25%. Dan risiko bahawa kanak-kanak itu akan menjadi pembawa gen ini (tetapi tidak akan mempunyai sebarang gejala) dianggarkan sebanyak 50%.
Dalam kes pewarisan berkaitan X (apabila gen bermutasi berada pada kromosom X wanita), terdapat risiko 50% bahawa ibu akan mewariskan gen kepada anaknya, yang akan mengembangkan CMT. Penyakit ini mungkin tidak berlaku apabila anak perempuan dilahirkan, tetapi anak lelaki (cucu) anak perempuan mungkin mewarisi gen yang rosak, dan penyakit itu akan berkembang.
Patogenesis
Dalam mana-mana jenis penyakit Charcot-Marie-Tooth, patogenesisnya disebabkan oleh anomali keturunan saraf periferi: motor (pergerakan) dan deria (sensitif).
Sekiranya jenis CMT mengalami demyelinating, maka kemusnahan atau kecacatan sarung mielin yang melindungi akson saraf periferi membawa kepada kelembapan dalam penghantaran impuls saraf dalam sistem saraf periferi - antara otak, otot dan organ deria.
Dalam jenis axonal penyakit ini, akson itu sendiri terjejas, yang memberi kesan negatif kepada kekuatan isyarat saraf, yang tidak mencukupi untuk rangsangan penuh otot dan organ deria.
Baca juga:
Bagaimanakah sindrom Charcot-Marie-Tooth dihantar? Gen yang rosak boleh diwarisi secara autosomal dominan atau autosomal resesif.
Jenis yang paling biasa, warisan dominan autosomal, berlaku apabila terdapat satu salinan gen yang bermutasi (dibawa oleh salah seorang ibu bapa). Dan kebarangkalian untuk menyampaikan CMT kepada setiap anak yang dilahirkan dianggarkan sebanyak 50%. [ 4 ]
Dengan pewarisan resesif autosomal, dua salinan gen yang rosak (satu daripada setiap ibu bapa yang tidak menunjukkan sebarang tanda penyakit) diperlukan untuk penyakit itu berkembang.
Dalam 40-50% kes, demielinasi keturunan dominan autosomal berlaku, iaitu jenis CMT 1; dalam 12-26% kes, CMT aksonal, iaitu jenis 2. Dan dalam 10-15% kes, warisan berkaitan X diperhatikan. [ 5 ]
Gejala Penyakit Charcot-Marie-Tooth
Biasanya, tanda-tanda pertama penyakit ini mula muncul pada zaman kanak-kanak dan remaja dan secara beransur-ansur berkembang sepanjang hayat, walaupun sindrom itu mungkin diketahui kemudian. Gabungan gejala adalah berubah-ubah, dan kadar perkembangan penyakit, serta keparahannya, adalah mustahil untuk diramalkan.
Gejala biasa peringkat awal termasuk peningkatan keletihan umum; penurunan nada (kelemahan) otot kaki, buku lali dan tulang kering; kekurangan refleks. Ini menjadikan pergerakan kaki sukar dan membawa kepada dysbasia (gangguan berjalan) dalam bentuk kenaikan kaki yang lebih tinggi, selalunya dengan kerap tersandung dan jatuh. Tanda-tanda penyakit Charcot-Marie-Tooth pada kanak-kanak kecil mungkin termasuk kejanggalan yang ketara dan kesukaran berjalan tanpa ciri usia yang dikaitkan dengan kejatuhan kaki dua hala. Kecacatan kaki juga merupakan ciri: gerbang tinggi (kaki berongga) atau kaki rata yang teruk, jari kaki melengkung (berbentuk tukul).
Dalam kes berjalan di atas jari kaki terhadap latar belakang hipotonia otot, pakar neurologi mungkin mengesyaki bahawa kanak-kanak itu mempunyai jenis 4 CMT, di mana kanak-kanak mungkin tidak dapat berjalan pada masa remaja.
Apabila penyakit itu berlanjutan, atrofi otot dan kelemahan merebak ke bahagian atas, menjadikannya sukar untuk melakukan kemahiran motor halus dan melakukan tugas tangan biasa. Penurunan sensasi sentuhan dan keupayaan untuk merasakan panas dan sejuk, serta kebas pada kaki dan tangan, menunjukkan kerosakan pada akson saraf deria.
Dalam penyakit Charcot-Marie-Tooth jenis 3 dan 6 yang nyata pada zaman kanak-kanak, ataxia deria (gangguan koordinasi pergerakan dan keseimbangan), otot berkedut dan gegaran, kerosakan pada saraf muka, atrofi saraf optik dengan nystagmus, dan kehilangan pendengaran diperhatikan.
Pada peringkat seterusnya, mungkin terdapat gegaran (gegaran) yang tidak terkawal dan kekejangan otot yang kerap; masalah dengan pergerakan boleh membawa kepada perkembangan kesakitan: otot, sendi, neuropatik.
Komplikasi dan akibatnya
Penyakit Charcot-Marie-Tooth boleh mempunyai komplikasi dan akibat seperti:
- lebih kerap terseliuh dan patah tulang;
- kontraktur yang berkaitan dengan pemendekan otot dan tendon periartikular;
- scoliosis (kelengkungan tulang belakang);
- masalah pernafasan - apabila gentian saraf yang mempersarafi otot diafragma rosak:
- kehilangan keupayaan untuk bergerak secara bebas.
Diagnostik Penyakit Charcot-Marie-Tooth
Diagnosis termasuk pemeriksaan klinikal, anamnesis (termasuk sejarah keluarga), pemeriksaan neurologi dan sistemik.
Ujian dilakukan untuk memeriksa julat pergerakan, sensitiviti dan refleks tendon. Pengaliran saraf boleh dinilai menggunakan diagnostik instrumental - elektromiografi atau electroneuromyography. Ultrasound atau MRI juga mungkin diperlukan. [ 6 ]
Ujian genetik atau DNA untuk mengesan mutasi genetik yang paling biasa yang menyebabkan CMT dalam sampel darah adalah terhad kerana ujian DNA tidak tersedia pada masa ini untuk semua jenis penyakit. Untuk maklumat lanjut, lihat Ujian genetik
Dalam sesetengah kes, biopsi saraf periferi (biasanya saraf sural) dilakukan.
Diagnosis pembezaan
Diagnosis pembezaan termasuk neuropati periferi yang lain, distrofi otot Duchenne, sindrom myelopathic dan myasthenic, neuropati diabetik, myelopathies dalam sklerosis lateral berbilang dan amyotrophic, sindrom Guillain-Barré, trauma pada saraf peroneal dan atrofinya (termasuk apabila terjepit di antara cakera lumbar atau thalamus) sebagai kerosakan sampingan pada tulang belakang. kemoterapi (semasa rawatan dengan sitostatik seperti Vincristine atau Paclitaxel). [ 7 ]
Siapa yang hendak dihubungi?
Rawatan Penyakit Charcot-Marie-Tooth
Hari ini, rawatan untuk penyakit keturunan ini termasuk terapi senaman (bertujuan untuk menguatkan dan meregangkan otot); terapi pekerjaan (yang membantu pesakit dengan kelemahan otot di tangan); dan penggunaan alat ortopedik untuk memudahkan berjalan. Jika perlu, ubat penahan sakit atau antikonvulsan ditetapkan. [ 8 ]
Dalam kes kaki rata yang teruk, osteotomi boleh dilakukan, dan dalam kes ubah bentuk tumit, pembetulan pembedahan ditunjukkan - arthrodesis. [ 9 ]
Penyelidikan sedang dijalankan terhadap kedua-dua komponen genetik penyakit dan kaedah rawatannya. Penggunaan sel stem, hormon tertentu, lesitin atau asid askorbik masih belum membuahkan hasil yang positif.
Tetapi terima kasih kepada penyelidikan baru-baru ini, sesuatu yang baru mungkin muncul dalam rawatan penyakit Charcot-Marie-Tooth dalam masa terdekat. Oleh itu, sejak 2014, syarikat Perancis Pharnext telah berkembang, dan sejak pertengahan 2019, ujian klinikal telah dijalankan untuk ubat PXT3003 untuk rawatan CMT jenis 1 pada orang dewasa, yang menekan peningkatan ekspresi gen PMP22, meningkatkan myelinasi saraf periferi dan mengurangkan gejala neuromuskular.
Pakar dari syarikat perubatan Sarepta Therapeutics (USA) sedang berusaha mencipta terapi gen untuk penyakit Charcot-Marie-Tooth jenis 1. Terapi ini akan menggunakan virus berkaitan adeno (AAV) yang tidak berbahaya daripada genus Dependovirus dengan genom DNA untai tunggal linear, yang akan memindahkan gen NTF3 ke dalam badan, mengekod protein neurotrophin-3 (NT-3) yang diperlukan untuk berfungsi sel saraf Schwann.
Helixmith akan memulakan ujian klinikal terapi gen Engensis (VM202) yang dibangunkan Korea Selatan pada penghujung tahun 2020 untuk merawat gejala otot dalam jenis CMT 1. [ 10 ]
Pencegahan
Pencegahan CMT boleh menjadi kaunseling genetik ibu bapa masa depan, terutamanya jika seseorang dalam pasangan itu mempunyai sejarah keluarga penyakit itu. Walau bagaimanapun, kes mutasi gen titik de novo telah dikenalpasti, iaitu, jika tiada penyakit dalam sejarah keluarga.
Semasa kehamilan, kebarangkalian penyakit Charcot-Marie-Tooth pada anak masa depan boleh diperiksa dengan biopsi vilus chorionic (dari 10 hingga 13 minggu kehamilan), serta analisis cecair amniotik (pada 15-18 minggu).
Ramalan
Secara umum, prognosis untuk pelbagai jenis penyakit Charcot-Marie-Tooth bergantung pada keterukan klinikal, tetapi dalam semua kes penyakit itu berkembang dengan perlahan. Ramai pesakit mempunyai kecacatan, walaupun ini tidak mengurangkan jangka hayat.