Pakar perubatan artikel itu

Penerbitan baru
Sindrom Martin-Bell
Ulasan terakhir: 04.07.2025

Semua kandungan iLive disemak secara perubatan atau fakta diperiksa untuk memastikan ketepatan faktual sebanyak mungkin.
Kami mempunyai garis panduan sumber yang ketat dan hanya memautkan ke tapak media yang bereputasi, institusi penyelidikan akademik dan, apabila mungkin, dikaji semula kajian secara medis. Perhatikan bahawa nombor dalam kurungan ([1], [2], dan lain-lain) boleh diklik pautan ke kajian ini.
Jika anda merasakan bahawa mana-mana kandungan kami tidak tepat, ketinggalan zaman, atau tidak dipersoalkan, sila pilih dan tekan Ctrl + Enter.
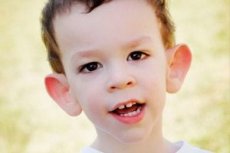
Sindrom Martin-Bell telah diterangkan pada tahun 1943 oleh doktor, selepas itu ia dinamakan. Penyakit ini adalah gangguan genetik yang terdiri daripada terencat akal. Pada tahun 1969, perubahan dalam kromosom X (kerapuhan pada lengan distal) ciri penyakit ini telah dikenalpasti. Pada tahun 1991, saintis menemui gen yang bertanggungjawab untuk perkembangan penyakit ini. Penyakit ini juga dipanggil "sindrom X rapuh". Kedua-dua kanak-kanak lelaki dan perempuan terdedah kepada penyakit ini, tetapi kanak-kanak lelaki lebih kerap (3 kali) terjejas.
Epidemiologi
Sindrom Martin-Bell adalah penyakit yang agak biasa: 0.3-1.0 daripada 1,000 lelaki menderita penyakit ini, dan 0.2-0.6 daripada 1,000 wanita. Selain itu, kanak-kanak dengan sindrom Martin-Bell dilahirkan di semua benua dengan kekerapan yang sama. Jelas sekali, kewarganegaraan, warna kulit, bentuk mata, keadaan hidup, dan kesejahteraan orang ramai tidak menjejaskan kejadian penyakit ini. Kekerapan kejadiannya hanya setanding dengan kekerapan sindrom Down (1 penyakit daripada 600-800 bayi baru lahir). Seperlima pembawa gen lelaki yang diubah adalah sihat, tidak mempunyai kelainan klinikal atau gen, selebihnya mempunyai tanda-tanda terencat akal dari bentuk ringan hingga teruk. Di kalangan pembawa wanita, lebih sedikit daripada satu pertiga adalah sakit.
Sindrom Fragile X menjejaskan kira-kira 1 dalam 2,500–4,000 lelaki dan 1 dalam 7,000–8,000 perempuan. Prevalens pembawa di kalangan wanita dianggarkan 1 dalam 130-250; kelaziman pembawa dalam kalangan lelaki dianggarkan 1 dalam 250–800.
Punca Sindrom Martin-Bell
Sindrom Martin-Bell berkembang disebabkan oleh pemberhentian sepenuhnya atau sebahagian daripada pengeluaran protein tertentu oleh badan. Ini berlaku kerana kekurangan tindak balas daripada gen FMR1, yang dilokalkan dalam kromosom X. Mutasi berlaku akibat daripada penstrukturan semula gen daripada varian struktur yang tidak stabil bagi keadaan gen (alel), dan bukan dari awal lagi. Penyakit ini ditularkan hanya melalui talian lelaki, dan lelaki itu mungkin tidak semestinya sakit. Pembawa lelaki menghantar gen kepada anak perempuan mereka dalam bentuk yang tidak berubah, jadi terencat akal mereka tidak jelas. Dengan penghantaran selanjutnya gen dari ibu kepada anak-anaknya, gen bermutasi, dan semua tanda ciri penyakit ini muncul.
Patogenesis
Patogenesis sindrom Martin-Bell adalah berdasarkan mutasi radas gen, yang membawa kepada menghalang pengeluaran protein FMR, protein yang penting untuk badan, terutamanya dalam neuron, dan terdapat dalam pelbagai tisu. Penyelidikan menunjukkan bahawa protein FMR terlibat secara langsung dalam proses mengawal selia terjemahan yang berlaku dalam tisu otak. Ketiadaan protein ini atau pengeluarannya yang terhad oleh badan membawa kepada terencat akal.
Dalam patogenesis penyakit, hipermetilasi gen dianggap sebagai gangguan utama, tetapi masih belum mungkin untuk mengenal pasti secara pasti mekanisme perkembangan gangguan ini.
Pada masa yang sama, heterogenitas lokus patologi juga ditemui, yang dikaitkan dengan polyalelisme, serta polylocus. Kehadiran varian alel perkembangan penyakit ditentukan, yang disebabkan oleh kewujudan mutasi titik, serta pemusnahan gen jenis FMRL.
Pesakit juga mempunyai 2 triplet rapuh yang sensitif kepada asid folik, terletak 300 kb, serta 1.5-2 juta bp daripada triplet rapuh yang mengandungi gen FMR1. Mekanisme mutasi yang berlaku dalam gen FRAXE dan FRAXF (ia dikenal pasti dalam kembar tiga rapuh yang disebutkan di atas) berkaitan dengan mekanisme gangguan dalam sindrom Martin Bell. Mekanisme ini disebabkan oleh penyebaran ulangan GCC dan CGG, yang menyebabkan metilasi pulau-pulau yang dipanggil CpG. Sebagai tambahan kepada bentuk klasik patologi, terdapat juga 2 jenis jarang yang berbeza disebabkan oleh pengembangan ulangan trinukleotida (dalam meiosis lelaki dan wanita).
Didapati bahawa dalam bentuk klasik sindrom, pesakit tidak mempunyai protein nukleositoplasma khas jenis FMR1, yang melaksanakan fungsi mengikat pelbagai mRNA. Di samping itu, protein ini menggalakkan pembentukan kompleks yang membantu menjalankan proses terjemahan di dalam ribosom.
Gejala Sindrom Martin-Bell
Bagaimana untuk mengenali penyakit pada kanak-kanak? Apakah tanda-tanda pertama? Pada bulan-bulan pertama kehidupan kanak-kanak, adalah mustahil untuk mengenali gejala Martin-Bell, kecuali kadang-kadang penurunan dalam nada otot diperhatikan. Selepas setahun, gambaran klinikal penyakit itu lebih jelas: kanak-kanak mula berjalan dan bercakap lewat, kadang-kadang ucapan tidak hadir sepenuhnya. Dia hiperaktif, melambai-lambaikan tangannya secara rawak, takut kepada orang ramai dan bunyi bising, degil, terdapat kemarahan yang tajam, ketidakstabilan emosi, sawan epilepsi berlaku, tidak membuat hubungan mata. Pada pesakit dengan sindrom Martin-Bell, penyakit ini juga diberikan oleh penampilan mereka: telinga menonjol dan besar, dahi berat, muka memanjang, dagu menonjol, strabismus, tangan dan kaki lebar. Mereka juga dicirikan oleh gangguan endokrin: selalunya berat badan besar, obesiti, buah zakar besar pada lelaki, akil baligh awal.

Di kalangan pesakit sindrom Martin-Bell, tahap kecerdasan sangat berbeza: daripada terencat akal ringan kepada kes yang teruk. Jika seseorang normal mempunyai kecerdasan kecerdasan (IQ) 100 secara purata, dan seorang genius mempunyai 130, maka orang yang terdedah kepada penyakit itu mempunyai 35-70.
Semua gejala klinikal patologi boleh dicirikan oleh triad manifestasi utama:
- oligofrenia (IQ ialah 35-50);
- dysmorphophobia (telinga yang menonjol dan prognatisme diperhatikan);
- makroorkidisme, yang muncul selepas permulaan akil baligh.
Kira-kira 80% pesakit juga mengalami prolaps injap bikuspid.
Walau bagaimanapun, bentuk penuh sindrom menunjukkan dirinya hanya dalam 60% daripada semua pesakit. Dalam 10%, hanya terencat akal yang dikesan, dan selebihnya, penyakit itu berkembang dengan kombinasi gejala yang berbeza.
Antara tanda-tanda awal penyakit, yang muncul pada usia awal:
- kanak-kanak yang sakit menunjukkan terencat akal yang ketara berbanding dengan perkembangan rakan sebaya yang lain;
- gangguan perhatian dan tumpuan;
- kedegilan yang kuat;
- kanak-kanak mula berjalan dan bercakap agak lewat;
- hiperaktif dan gangguan perkembangan pertuturan diperhatikan;
- serangan kemarahan yang sangat kuat dan tidak terkawal;
- mutisme mungkin berkembang - ini adalah kekurangan pertuturan yang lengkap pada kanak-kanak;
- bayi mengalami kebimbangan sosial dan mampu panik kerana bunyi yang kuat atau sebarang bunyi kuat yang lain;
- kanak-kanak itu melambai tangannya tanpa kawalan dan kelam kabut;
- rasa malu diperhatikan, kanak-kanak itu takut berada di tempat yang sesak;
- kemunculan pelbagai idea obsesif, keadaan emosi yang tidak stabil;
- Bayi mungkin keberatan untuk membuat hubungan mata dengan orang lain.
Pada orang dewasa, gejala patologi berikut diperhatikan:
- penampilan khusus: muka yang memanjang dengan dahi yang berat, telinga yang besar menonjol, dagu yang menonjol dengan kuat;
- kaki rata, otitis dan strabismus;
- akil baligh berlaku agak awal;
- obesiti boleh berkembang;
- Selalunya, kecacatan jantung diperhatikan dalam sindrom Martin-Bell;
- pada lelaki, pembesaran buah zakar diperhatikan;
- artikulasi sendi menjadi sangat mudah alih;
- berat dan ketinggian meningkat secara mendadak.
Diagnostik Sindrom Martin-Bell
Untuk mendiagnosis sindrom Martin Bell, anda perlu menghubungi pakar genetik yang berkelayakan. Diagnosis dibuat selepas ujian genetik tertentu yang membolehkan anda mengenal pasti kromosom yang rosak.
Ujian
Pada peringkat awal penyakit ini, kaedah sitogenetik digunakan, di mana serpihan bahan selular diambil dari pesakit, yang kemudiannya asid folik ditambah untuk mencetuskan perubahan dalam kromosom. Selepas tempoh masa tertentu, kawasan kromosom dikenal pasti di mana terdapat penipisan yang ketara - ini adalah tanda kehadiran sindrom X rapuh.
Walau bagaimanapun, ujian ini tidak sesuai untuk diagnosis pada peringkat akhir penyakit ini, kerana ketepatannya dikurangkan oleh penggunaan meluas multivitamin yang mengandungi asid folik.
Diagnostik bersepadu sindrom Martin-Bell ialah pemeriksaan genetik molekul, yang terdiri daripada menentukan bilangan ulangan trinukleotida yang dipanggil dalam gen.
 [ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ]
[ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ]
Diagnostik instrumental
Kaedah diagnostik instrumental yang sangat spesifik ialah PCR (tindak balas rantai polimerase), yang membolehkan seseorang mengkaji struktur sisa asid amino yang terkandung dalam kromosom X dan dengan itu menentukan kehadiran sindrom Martin Bell.
Terdapat juga kaedah diagnostik patologi yang berasingan, malah lebih spesifik - gabungan PCR dan pengesanan menggunakan elektroforesis kapilari. Kaedah ini sangat tepat dan mengesan patologi kromosom pada pesakit dengan kegagalan ovari primer, serta sindrom ataxic.
Kehadiran kecacatan boleh ditentukan selepas melakukan diagnostik pada EEG. Pesakit dengan penyakit ini mempunyai aktiviti otak bioelektrik yang serupa.
Ujian apa yang diperlukan?
Diagnosis pembezaan
Kaedah yang berbeza yang membantu untuk mengesyaki sindrom termasuk:
- klinikal - 97.5% pesakit mempunyai tanda-tanda terencat akal yang jelas (sederhana atau mendalam); 62% mempunyai telinga besar yang menonjol; 68.4% mempunyai dagu dan dahi yang menonjol; 68.4% kanak-kanak lelaki mempunyai buah zakar yang membesar, 41.4% mempunyai keanehan pertuturan (kadar pertuturan tidak sekata, kelantangan tidak terkawal, dll.);
- sitogenik - darah diperiksa untuk kultur limfosit, bilangan sel dengan kromosom X yang rapuh setiap 100 sel yang dikaji ditentukan;
- Electroencephalography - perubahan dalam impuls elektrik otak yang khusus untuk sindrom Martin-Bell direkodkan.
Siapa yang hendak dihubungi?
Rawatan Sindrom Martin-Bell
Dalam rawatan pesakit dewasa, antidepresan dengan psikostimulan digunakan. Proses terapi dadah sentiasa dipantau oleh pakar psikologi dan psikiatri. Di samping itu, prosedur suntikan mikro dengan ubat-ubatan seperti Cerebrolysin (atau derivatifnya), serta sitomedines (seperti Solcoseryl atau Lidase) dilakukan di klinik swasta.
Dalam perkembangan sindrom ataxic, ubat penipisan darah dan nootropik digunakan. Di samping itu, campuran asid amino dan angioprotectors ditetapkan. Wanita yang mengalami kegagalan ovari primer ditetapkan rawatan pembetulan menggunakan ubat herba dan estrogen.
Antagonis reseptor glutamin juga digunakan dalam rawatan.
Secara tradisinya, rawatan sindrom Martin-Bell melibatkan penggunaan ubat-ubatan yang menjejaskan gejala penyakit, tetapi bukan puncanya. Terapi ini melibatkan preskripsi antidepresan, neuroleptik, dan psikostimulan. Tidak semua ubat ditunjukkan untuk digunakan pada kanak-kanak, jadi senarai ubat agak terhad. Neuroleptik yang boleh digunakan selepas 3 tahun (usia paling awal untuk preskripsi mereka) termasuk haloperidol dalam titisan dan tablet, klorpromazine dalam larutan, dan periciazine dalam titisan. Oleh itu, dos haloperidol untuk kanak-kanak dikira bergantung kepada berat badan. Bagi orang dewasa, dos ditetapkan secara individu. Ia diambil secara lisan, bermula dengan 0.5-5 mg 2-3 kali sehari, kemudian dos secara beransur-ansur meningkat kepada 10-15 mg. Apabila penambahbaikan berlaku, mereka beralih kepada dos yang lebih rendah untuk mengekalkan keadaan yang dicapai. Dalam kes pergolakan psikomotor, 5-10 mg ditetapkan secara intramuskular atau intravena, beberapa pengulangan mungkin selepas 30-40 minit. Dos harian tidak boleh melebihi 100 mg. Kesan sampingan dalam bentuk loya, muntah, kekejangan otot, peningkatan tekanan, aritmia, dan lain-lain adalah mungkin. Warga emas harus mengambil langkah berjaga-jaga khusus, kerana kes-kes henti jantung mengejut telah didaftarkan, dan tardive dyskinesia (pergerakan sukarela) mungkin berlaku.
Antidepresan meningkatkan aktiviti struktur otak, melegakan kemurungan, ketegangan, dan meningkatkan mood. Ubat-ubatan ini, disyorkan untuk digunakan dari umur 5-8 tahun untuk sindrom Martin-Bell, termasuk clomipromine, sertraline, fluoxetine, dan fluvoxamine. Oleh itu, fluoxetine diambil secara lisan semasa makan 1-2 kali (sebaik-baiknya pada separuh pertama hari), bermula dengan 20 mg sehari, meningkat kepada 80 mg jika perlu. Orang tua tidak disyorkan dos yang lebih tinggi daripada 60 mg. Kursus rawatan ditentukan oleh doktor, tetapi tidak lebih daripada 5 minggu.
Kesan sampingan yang mungkin: pening, kebimbangan, tinnitus, hilang selera makan, takikardia, edema, dll. Berhati-hati diperlukan apabila memberi preskripsi kepada orang tua, mereka yang mempunyai penyakit kardiovaskular dan diabetes.
Psikostimulan adalah ubat psikotropik yang digunakan untuk meningkatkan persepsi rangsangan luar: mereka menajamkan pendengaran, tindak balas tindak balas, dan penglihatan.
Diazepam ditetapkan sebagai penenang untuk neurosis, kebimbangan, sawan epilepsi, dan sawan. Ia diambil secara lisan, intravena, intramuskular, rektum (ke dalam rektum). Ia ditetapkan secara individu, bergantung kepada keparahan penyakit, dengan dos terkecil 5-10 mg, setiap hari - 5-20 mg. Tempoh rawatan adalah 2-3 bulan. Bagi kanak-kanak, dos dikira dengan mengambil kira berat badan dan ciri-ciri individu. Kesan sampingan termasuk kelesuan, sikap tidak peduli, mengantuk, loya, sembelit. Ia berbahaya untuk digabungkan dengan alkohol, ketagihan kepada dadah adalah mungkin.
Dalam rawatan sindrom Martin-Bell, terdapat kes peningkatan dalam keadaan dan dengan pengenalan ubat-ubatan yang diperbuat daripada bahan haiwan (otak): cerebrolysate, cerebrolysin, cerebrolysate-M. Komponen utama ubat-ubatan ini adalah peptida yang menggalakkan pengeluaran protein dalam neuron, dengan itu mengisi semula protein yang hilang. Cerebrolysin ditadbir sebagai jet 5-10 ml, kursus rawatan terdiri daripada 20-30 suntikan. Ubat ini ditetapkan kepada kanak-kanak dari umur satu tahun, diberikan secara intramuskular setiap hari 1-2 ml selama sebulan. Sesi pentadbiran berulang adalah mungkin. Kesan sampingan dalam bentuk demam, kontraindikasi untuk wanita hamil.
Terdapat percubaan untuk merawat penyakit dengan asid folik, tetapi hanya aspek tingkah laku yang bertambah baik (tahap pencerobohan dan hiperaktif menurun, pertuturan bertambah baik), dan tiada apa yang berubah pada tahap intelektual. Untuk memperbaiki keadaan penyakit, asid folik ditetapkan, kaedah fisioterapi, terapi pertuturan, pembetulan pedagogi dan sosial ditunjukkan.
Persediaan litium juga dianggap berkesan, kerana ia membantu meningkatkan penyesuaian pesakit kepada persekitaran sosial, serta aktiviti kognitif. Selain itu, mereka juga mengawal tingkah lakunya dalam masyarakat.
Penggunaan herba untuk sindrom Martin-Bell adalah mungkin sebagai antidepresan. Herba yang membantu melegakan ketegangan, kebimbangan dan meningkatkan tidur termasuk valerian, pudina, thyme, wort St. John dan chamomile. Infusi disediakan seperti berikut: untuk 1 sudu teh herba kering, anda memerlukan segelas air mendidih, rebusan diselitkan selama sekurang-kurangnya 20 minit, diambil terutamanya pada waktu malam sebelum tidur atau pada sebelah petang. Satu sudu madu akan menjadi tambahan yang baik kepada mereka.
Rawatan fisioterapi
Untuk menghapuskan manifestasi neurologi, prosedur fisioterapeutik khas dijalankan - seperti latihan di kolam renang, kelonggaran otot dan akupunktur.
Rawatan pembedahan
Peringkat penting rawatan juga dianggap sebagai kaedah pembedahan plastik - operasi yang membantu meningkatkan penampilan pesakit. Pembedahan plastik pada anggota badan dan aurikel dilakukan, serta alat kelamin. Pembetulan ginekomastia dengan epispadia juga dilakukan, serta kecacatan lain dalam penampilan.
Pencegahan
Satu-satunya kaedah untuk mencegah penyakit ini adalah pemeriksaan pranatal wanita hamil. Terdapat pemeriksaan khas yang membolehkan pengesanan awal patologi, selepas itu disyorkan untuk menamatkan kehamilan. Sebagai alternatif, IVF digunakan, yang boleh membantu kanak-kanak untuk mewarisi kromosom X yang sihat.
Pencegahan pesakit bergantung kepada sama ada mutasi gen telah timbul semula atau telah diwarisi. Untuk ini, diagnostik genetik molekul dijalankan. Fakta bahawa ujian tidak mendedahkan "kromosom X rapuh" dalam saudara-mara bercakap memihak kepada "kesegaran" mutasi, yang bermaksud risiko mempunyai anak dengan sindrom Martin-Bell adalah sangat kecil. Dalam keluarga di mana terdapat orang yang sakit, ujian akan membantu untuk mengelakkan kes berulang.
Ramalan
Prognosis untuk sindrom Martin-Bell adalah baik untuk kehidupan, tetapi bukan untuk pemulihan. Jangka hayat bergantung kepada keparahan penyakit dan kecacatan yang berkaitan. Pesakit boleh menjalani kehidupan normal. Dalam bentuk sindrom Martin-Bell yang teruk, pesakit berisiko hilang upaya sepanjang hayat.
Jangka hayat
Sindrom Martin Bell tidak mempunyai kesan negatif yang serius terhadap kesihatan, jadi jangka hayat kebanyakan orang yang telah didiagnosis dengan patologi ini tidak berbeza daripada penunjuk standard.