Pakar perubatan artikel itu

Penerbitan baru
Nannisme pituitari (dwarfisme)
Ulasan terakhir: 12.07.2025

Semua kandungan iLive disemak secara perubatan atau fakta diperiksa untuk memastikan ketepatan faktual sebanyak mungkin.
Kami mempunyai garis panduan sumber yang ketat dan hanya memautkan ke tapak media yang bereputasi, institusi penyelidikan akademik dan, apabila mungkin, dikaji semula kajian secara medis. Perhatikan bahawa nombor dalam kurungan ([1], [2], dan lain-lain) boleh diklik pautan ke kajian ini.
Jika anda merasakan bahawa mana-mana kandungan kami tidak tepat, ketinggalan zaman, atau tidak dipersoalkan, sila pilih dan tekan Ctrl + Enter.
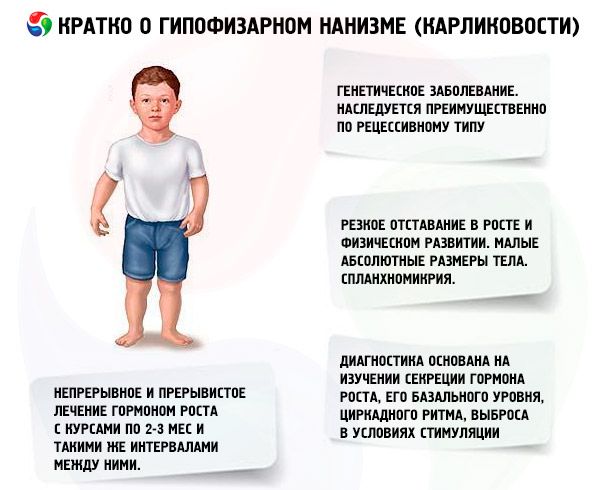
Istilah "dwarfisme pituitari" (dari nanos Yunani - kerdil; syn.: dwarfisme, nanosomia, microsomia) dalam erti kata mutlak bermaksud penyakit, manifestasi utamanya adalah keterlambatan tajam dalam pertumbuhan, yang dikaitkan dengan pelanggaran rembesan hormon pertumbuhan oleh kelenjar pituitari anterior.
Dalam erti kata yang lebih luas, dwarfisme adalah gangguan pertumbuhan dan perkembangan fizikal, kejadiannya boleh disebabkan bukan sahaja oleh kekurangan mutlak atau relatif hormon somatotropik akibat patologi kelenjar pituitari itu sendiri, tetapi juga oleh gangguan peraturan hipotalamus (cerebral) fungsinya, kecacatan dalam biosintesis hormon somatotropik ini, dan gangguan kepekaan hormon somatotropik ini.
 [
[ Punca kerdil
Kebanyakan bentuk dwarfisme pituitari adalah penyakit genetik. Yang paling biasa ialah kerdil panhypopituitary, yang diwarisi terutamanya dalam cara resesif. Diandaikan bahawa terdapat 2 jenis penghantaran bentuk patologi ini - autosomal dan melalui kromosom X. Dalam bentuk dwarfisme ini, bersama-sama dengan kecacatan dalam rembesan hormon somatotropik, rembesan gonadotropin dan hormon perangsang tiroid paling kerap terganggu. Rembesan ACTH kurang kerap terganggu dan sedikit sebanyak. Kajian fungsional dengan hormon pelepas, termasuk hormon pelepas somatotropin sintetik (terdiri daripada 29, 40 dan 44 sisa asid amino), serupa dengan polipeptida pankreas, telah menunjukkan bahawa kebanyakan pesakit ini mempunyai patologi pada tahap hipotalamus, dan kekurangan kelenjar pituitari anterior adalah sekunder. Patologi utama kelenjar pituitari itu sendiri kurang biasa.
Dwarfisme genetik dengan kekurangan hormon pertumbuhan terpencil, dengan aktiviti biologi terjejas dan kepekaan terhadapnya, ditemui secara sporadis di Rusia dan negara jiran. Ia lebih biasa di benua Amerika, di negara-negara Timur Dekat dan Timur Tengah, dan di Afrika. Berdasarkan hasil kajian kandungan darah hormon somatotropik dan kepekaan pesakit terhadap hormon somatotropik eksogen, tahap insulin imunoreaktif (IRI), faktor pertumbuhan seperti insulin (IGF) jenis I (somatomedin C) dan jenis II, dan tindak balas IGF-1 terhadap rawatan dengan persediaan hormon somatotropik, pelbagai varian jenis kerdil yang serupa secara klinikal telah dikenal pasti.
Baru-baru ini, patogenesis kerdil Laron, yang disebabkan oleh kekurangan IRF-1 dan IRF-II, telah diuraikan, serta patogenesis kerdil dalam pigmi Afrika, yang dikaitkan dengan kekurangan yang pertama.
Pada tahun 1984, varian baru kerdil pseudo-pituitari dengan tahap hormon somatotropik yang tinggi dan IGF-1 telah diterangkan; genesis dwarfisme dijelaskan oleh kecacatan pada reseptornya, yang dibuktikan dengan penurunan mendadak dalam pengikatan fibroblas kulit kepada IGF-1.
Perlu ditekankan bahawa dalam keadaan moden, dengan kehadiran keluarga kecil, banyak kes penyakit terpencil ("idiopatik", sporadis) juga boleh menjadi genetik.
Dalam analisis 350 sejarah kes, etiologi dwarfisme tidak jelas dalam 228 pesakit (65.2%). Kumpulan ini termasuk pesakit daripada 57 keluarga dengan kejadian dwarfisme berulang (2-4 kes setiap keluarga), yang menyumbang 28% daripada semua pesakit. Dalam 77% keluarga dengan bentuk dwarfisme yang tidak jelas (kebanyakannya genetik), hubungan yang tidak dapat dinafikan dengan warisan ketiadaan faktor Rh telah ditubuhkan. Pengagihan faktor Rh dalam keluarga pesakit dengan kerdil berbeza daripada yang diperhatikan dalam konflik Rh antara ibu dan janin dan, sebagai peraturan, tidak disertai oleh penyakit hemolitik bayi yang baru lahir (bapa mungkin Rh negatif, dan dalam kes heterozigositas ibu bapa untuk faktor Rh - kanak-kanak, dll.). Adalah mungkin untuk mengandaikan hubungan antara aktiviti gen yang bertanggungjawab untuk sintesis hormon somatotropik (atau hormon pelepas somatotropin) dan gen yang menentukan faktor Rh, terutamanya kerana kebanyakan bentuk dwarfisme dan ketiadaan faktor Rh adalah ciri resesif autosomal. Ini tidak menjelaskan kelangkaan relatif dwarfisme berbanding kekerapan individu Rh-negatif dalam populasi. Mungkin, beberapa faktor tambahan yang belum diketahui adalah penting, tetapi ciri-ciri taburan faktor Rh dalam keluarga pesakit dengan kekerdilan keluarga dan sporadis tidak mungkin berlaku secara tidak sengaja.
Sekumpulan besar pesakit dengan dwarfisme (cerebral primer, cerebral-pituitari) adalah pesakit dengan pelbagai jenis patologi organik sistem saraf pusat yang timbul dalam utero atau pada zaman kanak-kanak awal. Substrat anatomi yang menyebabkan patologi ini mungkin kekurangan atau ketiadaan kelenjar pituitari, distopia dalam patologi pembentukan sella turcica, degenerasi sista kelenjar pituitari, atrofinya akibat mampatan oleh tumor (craniopharyngioma, chromophobe adenoma, meningioma, glioma). Dwarfisme mungkin disebabkan oleh kecederaan traumatik pada kawasan hypothalamic-pituitari (intrauterine, kelahiran atau postnatal), yang sering berlaku dalam kehamilan berganda, serta semasa bersalin dalam sungsang, pembentangan kaki atau dalam kedudukan melintang dengan putaran pada kaki (ini adalah mekanisme bersalin pada lebih daripada 1/3 pesakit dengan kerdil). Kerosakan berjangkit dan toksik adalah penting (jangkitan virus intrauterin, batuk kering, sifilis, malaria, toksoplasmosis; penyakit pada usia awal, sepsis neonatal, meningo- dan arachnoencephalitis, dll.). Proses ini boleh merosakkan kelenjar pituitari itu sendiri, pusat hipotalamus yang mengawal fungsinya, dan mengganggu sambungan fungsi normal dalam sistem saraf pusat.
Lesi janin dalam rahim boleh membawa kepada kelahiran pesakit dengan "kerdil sejak lahir" dengan rembesan normal hormon pertumbuhan (kerdil primordial serebrum, microcephaly, kerdil Silver-Russell dengan hemi-asimetri badan dan tahap gonadotropin yang tinggi, dsb.).
Faktor tambahan yang memburukkan lagi pelanggaran perkembangan fizikal dalam kerdil mungkin pemakanan yang tidak mencukupi, tidak seimbang dari segi bahan penting (kekurangan protein) dan mikroelemen (kekurangan zink), dan keadaan persekitaran yang tidak menggalakkan, serta pelbagai penyakit kronik, seperti glomerulonephritis, di mana azotemia menjejaskan aktiviti reseptor hati atau penurunan sel hati, menyebabkan metabolisme sel-sel somatodin yang tidak baik, serta pelbagai penyakit kronik, seperti glomerulonephritis, di mana azotemia menjejaskan aktiviti reseptor hati atau sel-sel hati, menyebabkan simatodin. sirosis hati, apabila pembentukan somatomedin terjejas.
Patogenesis
Pada kebanyakan pesakit dengan kerdil pituitari, perubahan tidak terhad kepada patologi rembesan hormon somatotropik dan kepekaan kepadanya, tetapi meluas ke hormon tropik lain kelenjar pituitari, yang menyebabkan pelbagai kombinasi gangguan endokrin dan metabolik.
Dalam kekurangan hormon somatotropik terpencil, perubahan morfologi dalam kelenjar pituitari telah kurang dikaji. Dalam kes yang dikaji, gangguan patologi jarang ditemui (craniopharyngioma atau hyperostosis tulang tengkorak). Dalam jenis dwarfisme ini, keterbelakangan kongenital sel peptidergik atau kecacatan dalam sistem neurotransmitter dalam hipotalamus mungkin diperhatikan. Dalam kes sedemikian, dwarfisme boleh digabungkan dengan displasia atau hipoplasia saraf optik. Sista intrasellar, tumor pituitari dan hipotalamus membawa kepada kekurangan STH, menyebabkan mampatan tisu pituitari, khususnya somatotrof.
Dwarfisme dicirikan oleh penipisan tulang, terutamanya disebabkan oleh lapisan kortikal, pembezaan tertunda dan pengerasan rangka. Organ dalaman adalah hipoplastik, kadang-kadang atropik, dan otot kurang berkembang.
Gejala kerdil
Kelewatan mendadak dalam pertumbuhan dan perkembangan fizikal adalah manifestasi utama kerdil pituitari. Pesakit dilahirkan dengan berat dan panjang badan yang normal dan mula ketinggalan dalam pertumbuhan dari umur 2-4 tahun.
Sebelum kemunculan rawatan aktif untuk nanisme, kerdil dianggap sebagai mereka yang mempunyai ketinggian kurang daripada 120 cm untuk wanita dan 130 cm untuk lelaki. Pada masa ini, ketinggian kerdil berbeza dengan sekurang-kurangnya 2-3 sisihan sigma daripada norma jadual purata untuk jantina, umur atau populasi tertentu. Terdapat juga kaedah untuk penilaian grafik ketinggian berdasarkan keluk taburan Gaussian. Dalam kes ini, kerdil mengikut ketinggian termasuk dalam kumpulan yang merangkumi bilangan minimum individu dalam populasi yang sepadan dengan ketinggalan terbesar daripada norma pertumbuhan purata.
Dwarfisme pituitari dicirikan bukan sahaja oleh saiz badan mutlak yang kecil, tetapi juga oleh pertumbuhan tahunan yang kecil dan dinamik pembangunan fizikal. Binaan badan adalah berkadar, tetapi perkadaran badan pesakit adalah tipikal zaman kanak-kanak. Kulitnya pucat, selalunya dengan warna kekuningan, kering, yang disebabkan oleh kekurangan tiroid mutlak atau relatif, kadang-kadang sianosis diperhatikan - "marbling" kulit. Pada pesakit yang tidak dirawat, kulit yang kelihatan tua dan berkedut (geroderma) muncul lebih awal. Ini disebabkan oleh ketidakcukupan tindakan anabolik STH dan perubahan perlahan generasi selular.
Rambut di kepala mungkin normal atau kering, nipis, rapuh; bulu mata panjang adalah tipikal. Pertumbuhan rambut sekunder selalunya tidak hadir. Saiz sella turcica pada kebanyakan pesakit dengan kerdil (70-75%) tidak berubah, tetapi sella sering mengekalkan bentuk kebudak-budakan "bujur berdiri", mempunyai belakang "juvana" yang luas, sinus tulang sphenoid tertinggal dari segi pneumatisasi. Walau bagaimanapun, terdapat pesakit dengan sella turcica yang membesar, yang merupakan tanda tumor; dengan kawasan kalsifikasi pada latar belakangnya atau di kawasan pintu masuk (dengan craniopharyngioma, kesan sisa neuroinfection) atau penurunannya (tanda-tanda keterbelakangan, saiz kecil kelenjar pituitari). Gejala hipertensi intrakranial diperhatikan: penipisan tulang peti besi tengkorak, peningkatan corak vaskular, kehadiran kesan jari, dsb. Tanda paling penting bagi kerdil pituitari ialah kelewatan dalam masa pembezaan dan pengerasan rangka. Ciri-ciri sistem pergigian juga berkait rapat dengan pembezaan rangka: penggantian lewat gigi susu dicatatkan. Kelewatan terbesar dalam pembangunan sistem rangka diperhatikan pada pesakit dengan dwarfisme dengan kekurangan seksual dan hipotiroidisme.
Alat kelamin kebanyakan pesakit sangat kurang berkembang, walaupun kecacatan jarang berlaku. Kami memerhatikan cryptorchidism dalam 5.8% pesakit lelaki. Kekurangan seksual disertai dengan keterbelakangan ciri-ciri seksual sekunder dan penurunan keinginan seksual, ketiadaan haid. Perkembangan seksual spontan yang normal hanya diperhatikan pada pesakit dengan kekurangan hormon pertumbuhan terpencil dan pada sesetengah pesakit dengan kerdil serebrum.
Kekurangan tiroid adalah gejala kerdil yang agak biasa. Perlu diingatkan bahawa manifestasi luaran hipotiroidisme tidak selalu mencerminkan keadaan fungsi sebenar kelenjar tiroid. Ini disebabkan oleh hipotiroidisme relatif akibat pelanggaran peralihan tiroksin (T 4 ) kepada triiodothyronine (T 3 ) dan pembentukan T 3 yang tidak aktif (boleh balik), yang merupakan ciri kekurangan somatotropik.
Fungsi adrenokortikotropik dalam kerdil pituitari berkurangan kurang kerap dan pada tahap yang lebih rendah daripada fungsi merangsang seks dan tiroid, dan dalam kebanyakan pesakit tidak memerlukan pembetulan khas.
Dalam kebanyakan kes, intelek tidak terjejas. Perubahan emosi dalam bentuk infantilisme mental ditemui; pada pesakit yang lebih tua tanpa gangguan intelek, neurosis reaktif kadang-kadang diperhatikan.
Dalam patologi serebrum organik, terutamanya yang bersifat tumor, kerdil boleh berlaku dengan gejala diabetes insipidus, hemianopsia bitemporal dan ketidakupayaan intelektual.
Kajian mengenai perkembangan aktiviti bioelektrik otak pada pesakit tanpa gejala organik sistem saraf pusat menunjukkan bahawa EEG mereka dicirikan oleh ciri-ciri ketidakmatangan, pemeliharaan jangka panjang voltan EEG "kekanak-kanakan" yang tinggi; ketidaksamaan irama alfa dalam amplitud dan kekerapan; peningkatan mendadak dalam kandungan θ- dan δ-irama yang perlahan, terutamanya dalam petunjuk hadapan dan tengah; tindak balas yang jelas terhadap hiperventilasi; anjakan dalam julat irama EEG mengikuti irama rangsangan cahaya ke arah frekuensi rendah (bukti penurunan dalam mobiliti berfungsi struktur saraf otak). Telah mendedahkan bahawa pada pesakit yang lebih tua, sifat tidak matang aktiviti elektrik otak adalah disebabkan oleh keterbelakangan seksual, dan pada pesakit dari semua kumpulan umur - hipotiroidisme.
Metabolisme karbohidrat pada pesakit dengan kerdil dicirikan oleh kecenderungan untuk mengurangkan tahap glukosa darah puasa, peningkatan semasa senaman fizikal, kekurangan insulin endogen, peningkatan kepekaan terhadap insulin eksogen dengan perkembangan keadaan hipoglikemik yang kerap. Yang terakhir ini dijelaskan terutamanya oleh tahap hormon kontra-insular yang tidak mencukupi dalam badan pesakit.
Organ dalaman menunjukkan splanchnomycria, iaitu pengurangan saiznya. Tiada perubahan fungsi dalam organ dalaman khusus untuk dwarfisme telah diterangkan. Hipotensi arteri dengan penurunan tekanan sistolik dan diastolik dan penurunan amplitud nadi sering diperhatikan. Bunyi jantung tersekat, murmur berfungsi pelbagai topik kedengaran disebabkan oleh perubahan trofik dalam miokardium dan gangguan autonomi. ECG dicirikan oleh voltan rendah (terutamanya dengan kehadiran hipotiroidisme), bradikardia sinus atau bradyarrhythmia; PCG menunjukkan penurunan dalam amplitud ton, ton tambahan, dan murmur berfungsi. Data oxygemometry menunjukkan hipoksemia (awal dan semasa melakukan senaman fizikal), dan hutang oksigen. Pesakit warga emas kadangkala mengalami hipertensi.
Diagnostik kerdil
Diagnosis dan diagnosis pembezaan dwarfisme adalah berdasarkan data anamnesis dan pemeriksaan klinikal, radiologi, makmal dan hormon yang komprehensif. Sebagai tambahan kepada saiz badan mutlak, untuk menilai pertumbuhan pesakit, defisit pertumbuhan ditentukan - perbezaan antara ketinggian pesakit dan norma puratanya untuk jantina dan umur yang sepadan; umur pertumbuhan - pematuhan ketinggian pesakit dengan piawaian tertentu; penunjuk sisihan ternormal
I = M - Mcp / δ, di mana M ialah ketinggian pesakit, Mcp ialah purata ketinggian normal untuk jantina dan umur tertentu, δ ialah sisihan segi empat sama dari Mcp; Saya kurang daripada 3 adalah tipikal nanisme, I lebih daripada 3 adalah tipikal gigantisme. Penunjuk ini boleh digunakan untuk menilai dinamik pembangunan.
Pemeriksaan sinar-X pesakit kerdil mendedahkan tanda-tanda hipertensi intrakranial, kesan sisa neuroinfection, kalsifikasi, dan craniosynostosis. Kajian tentang saiz, bentuk, dan struktur sella turcica dianggap sebagai penunjuk tidak langsung yang mencirikan saiz kelenjar pituitari. Salah satu manifestasi paling penting dalam keterlambatan pertumbuhan patologi adalah pelanggaran pembezaan rangka. Untuk menilai tahap kematangan rangka, umur tulang (radiografi) ditentukan, yang sepadan dengan pembezaan tisu tulang; kekurangan osifikasi ialah tahap ketinggalan osifikasi dari norma (dalam tahun), pekali osifikasi ialah hasil pembahagian umur tulang mengikut kronologi dan parameter lain.
Diagnostik moden kerdil adalah mustahil tanpa mengkaji rembesan hormon somatotropik, tahap basalnya, irama sirkadian, dan pelepasan di bawah rangsangan. Kebanyakan pesakit dengan kerdil pituitari dicirikan oleh pengurangan kandungan hormon somatotropik dalam serum darah. Apabila ditentukan oleh kaedah radioimunologi, ia adalah (mengikut pengarang yang berbeza) daripada (0.87±0.09) kepada (1.50±0.64) ng/ml, dengan purata norma (3.81±0.29) ng/ml. Kajian tentang irama harian (sirkadian) rembesan hormon somatotropik menunjukkan bahawa tahapnya pada orang yang sihat adalah maksimum semasa 2 jam pertama tidur dan pada 4-6 pagi. Dalam kerdil, kandungan hormon somatotropik dikurangkan pada jam-jam ini juga.
Untuk mengkaji rizab fungsi somatotropik, pelbagai perangsang digunakan, memeriksa kandungan hormon somatotropik sebelum dan selepas pengenalannya. Darah untuk kajian diambil setiap 30 minit selama 2-3 jam. Pembebasan hormon somatotropik selepas rangsangan dianggap normal sekurang-kurangnya sehingga 7-10 ng/ml, kadangkala ia mencapai 20-40 ng/ml. Jika tiada tindak balas dalam salah satu sampel, ujian ulangan dijalankan dengan perangsang lain. Kekurangan hormon somatotropik dianggap terbukti jika tiada pelepasan hormon somatotropik dalam 2-3 sampel berbeza.
Ujian rangsangan yang paling biasa digunakan ialah: dengan pentadbiran intravena 0.1 U (0.75-1.5 U) insulin setiap 1 kg berat badan pesakit dan pencapaian hipoglikemia (penurunan tahap glukosa darah sebanyak 50% berbanding tahap awal), hormon somatotropik serum ditentukan mengikut skema di atas. Sekiranya hipoglikemia teruk berkembang, ujian terganggu, dan pesakit diberi glukosa secara intravena. Ini adalah kaedah diagnostik klasik yang paling biasa.
TRH dalam dos 200-500 mcg secara intravena. Berkesan mengenal pasti rizab hormon, tidak menyebabkan komplikasi. Dalam kombinasi dengan ujian insulin, ia membolehkan seseorang menilai tahap kerosakan pada sistem hipotalamus-pituitari. Reaksi positif terhadap TRH jika tiada satu kepada hipoglikemia insulin menunjukkan keutuhan kelenjar pituitari dan kerosakan pada tahap hipotalamus, tindak balas negatif terhadap TRH dan hipoglikemia menunjukkan kerosakan pada kelenjar pituitari itu sendiri.
TRH, LH-RH pada dos 300 mcg secara intravena adalah serupa dengan yang sebelumnya.
SGH manusia ialah analog sintetik bagi sebatian aktif biologi yang diasingkan daripada tumor pankreas. Pada masa ini, terdapat 3 jenis SGH sintetik: dengan 29, 40 dan 44 sisa asid amino. Ia digunakan secara intravena dalam dos dari 1 hingga 3 μg / kg berat badan pesakit. Pembebasan STH diperhatikan 15-20 minit selepas pentadbiran, ujian lebih berkesan daripada yang lain dalam mendedahkan rizab hormon pertumbuhan endogen. Tindak balas SGH positif menunjukkan tahap kerosakan hipotalamus pada fungsi somatotropik dan kelenjar pituitari yang utuh; dengan asid amino (L-arginine monochloride, ornithine, tryptophan, glycine, leucine) secara intravena pada dos 0.25-0.5 g setiap 1 kg berat badan pesakit. Berkesan untuk mengkaji rizab SGH. Boleh menyebabkan reaksi alahan.
L-dopa secara lisan pada dos 250-500 mcg. Berkesan, diterima dengan baik oleh pesakit.
Ujian dengan glukagon, bromin ergocryptine (parlodel), lysine vasopressin, clonidine, dan beban ergometrik basikal berdos juga digunakan.
Kajian tentang keadaan fungsi somatotropik adalah perlu bukan sahaja untuk diagnosis dwarfisme, tetapi juga untuk pilihan kaedah terapi yang wajar, kerana rawatan dengan somatotropin adalah rasional hanya dalam kes kekurangan hormon pertumbuhan endogen.
Untuk diagnosis bentuk dwarfisme, sangat penting untuk mengkaji kandungan faktor pertumbuhan seperti insulin, atau somatomedin (terutama IGF-1, atau somatomedin C) - mediator tindakan hormon somatotropik pada tahap tisu. Adalah diketahui bahawa kandungan somatomedin C dalam dwarfisme dikurangkan, dan dalam acromegali - meningkat berbanding dengan norma. Bentuk dwarfisme yang diterangkan oleh Laron adalah sejenis penyakit dengan pengeluaran STH yang normal, tetapi dengan pelanggaran pembentukan IGF-1 dan IGF-II. Rawatan pesakit sedemikian dengan somatotropin adalah sia-sia.
Penunjuk tidak langsung fungsi somatotropik kelenjar pituitari adalah aktiviti fosfatase alkali dan kandungan fosforus tak organik dalam serum. Dalam keadaan hiposomatotropik, penunjuk ini dikurangkan. Dalam bentuk panhypopituitary dwarfism, rembesan gonadotropin, selalunya TSH, dikurangkan, yang disertai dengan penurunan yang sepadan dalam fungsi kelenjar seks (kekurangan androgen atau estrogen), kelenjar tiroid (penurunan tahap T3 , T4 , yodium terikat protein - PBI13, pengumpulan kelenjar adrenal oleh kelenjar adrenal). jumlah kortisol dan 17-OCS dalam plasma, perkumuhan 17-KC dan 17-OCS dalam air kencing, limfositosis).
Semua jenis kerdil genetik pituitari (hipothalamik-pituitari) dicirikan oleh penyakit berulang kanak-kanak dalam keluarga dengan warisan oleh autosomal resesif (lebih kerap) atau jenis dominan autosomal, terencat pertumbuhan dan perkembangan fizikal dari 2-4 tahun dengan lag sekurang-kurangnya 2-3 o dari norma pertumbuhan purata untuk jantina yang rendah, pertumbuhan spontan tahunan tertentu, osifikasi. Dengan tahap hormon somatotropik yang rendah (dalam 2-3 ujian merangsang di bawah 7 ng / ml), terapi dengan hormon somatotropik sangat berkesan (memberi peningkatan ketinggian sekurang-kurangnya 7 cm setahun). Dengan tahap hormon somatotropik yang normal atau tinggi (dengan ketidakaktifan biologinya), sensitiviti terhadap hormon boleh dipelihara. Tiada perubahan dalam kecerdasan diperhatikan
Dalam kekerdilan genetik dengan ketidakpekaan tisu kepada hormon somatotropik, gambaran klinikal adalah serupa dengan kekurangan hormon pertumbuhan terpencil, tetapi terapi somatotropin tidak berkesan. Dalam kumpulan ini, bentuk utama berikut boleh dibezakan mengikut tahap IRF: dengan kandungan normal (kecacatan reseptor IRF) dan dikurangkan - Dwarfisme jenis Laron (kekurangan IRF-1 dan IRF-II) dan jenis yang terdapat dalam pigmi Afrika (kekurangan IRF-1).
Dwarfisme serebrum dicirikan oleh penyakit terpencil dalam keluarga yang berkaitan dengan kerosakan intrauterin atau postnatal pada sistem saraf pusat, dengan kehadiran perubahan organik yang jelas dalam sistem saraf pusat, sering digabungkan dengan patologi organ visual, kehadiran diabetes insipidus, pemeliharaan fungsi gonadotropik, dan perubahan dalam kecerdasan.
Beberapa jenis disgenesis gonad dan agenesis disertai dengan perawakan pendek yang ketara, khususnya sindrom Shereshevsky-Turner dan bentuk "Turneroid" (mosaik) sindrom disgenesis testis. Kajian sitogenetik (kromatin seks, karyotype) membantu dalam diagnosis pembezaan, mendedahkan kecacatan kromosom, serta kecacatan ciri perkembangan somatik dan seksual, tahap normal atau peningkatan hormon somatotropik endogen, dan ketidakpekaan terhadap rawatan dengan somatotropin.
Antara gangguan endokrin yang berlaku dengan perawakan pendek, hipotiroidisme primer harus dikhususkan, disebabkan oleh hipoplasia kongenital atau aplasia kelenjar tiroid, distopianya, kecacatan enzim dalam biosintesis hormon tiroid, kerosakan autoimun awal pada kelenjar tiroid. Dalam semua keadaan ini, tanda-tanda hipotiroidisme dengan tahap TSH yang tinggi, penurunan T4 dan T3 dalam serum darah mendominasi . Dalam myxedema genesis autoimun, antibodi kepada tiroglobulin, mikrosomal dan pecahan nuklear tisu tiroid dikesan dalam darah, tahap hormon somatotropik adalah normal atau berkurangan. Kesan klinikal boleh dicapai dengan mengimbangi hanya hipotiroidisme.
Perawakan pendek disertai dengan perkembangan seksual pramatang dan sindrom adrenogenital akibat penutupan awal zon pertumbuhan; Penyakit Itsenko-Cushing, yang berlaku pada zaman kanak-kanak disebabkan oleh kesan perencatan glukokortikoid pada rembesan hormon somatotropik dan kesan kataboliknya; Sindrom Mauriac - perawakan pendek dan infantilisme pesakit dengan diabetes mellitus yang bergantung kepada insulin yang teruk.
Dwarfisme pituitari harus dibezakan daripada kelewatan somatogenik dalam perkembangan fizikal yang disebabkan oleh gangguan metabolik kronik (dalam penyakit hati, buah pinggang, saluran gastrousus), hipoksia kronik (dalam penyakit sistem kardiovaskular dan pernafasan, dalam anemia); dengan penyakit sistemik sistem muskuloskeletal (chondrodystrophy, osteogenesis tidak sempurna, penyakit exostosis), dsb.
Kerencatan pertumbuhan fungsional (perlembagaan) kadangkala diperhatikan dengan permulaan akil baligh yang lewat pada remaja yang kelihatan sihat; kami telah mendapati bahawa ia dikaitkan terutamanya dengan ketidakcukupan sementara aktiviti gonadotropik. Rembesan hormon somatotropik biasanya tidak terjejas atau berkurangan sedikit. Rangsangan gonadotropin boleh mempercepatkan perkembangan dan pertumbuhan seksual.
Perawakan pendek yang bersifat kekeluargaan harus dianggap sebagai varian perkembangan fisiologi.
Apa yang perlu diperiksa?
Bagaimana untuk memeriksa?
Ujian apa yang diperlukan?
Siapa yang hendak dihubungi?
Rawatan kerdil
Rawatan dwarfisme adalah proses yang panjang. Ini memaksa doktor untuk mengagihkan cara mempengaruhi pertumbuhan dari semasa ke semasa untuk mendapatkan kesan klinikal yang paling besar sambil memerhatikan 2 prinsip asas:
- anggaran maksimum perkembangan yang disebabkan oleh rawatan kepada keadaan fisiologi;
- menyelamatkan zon pertumbuhan epifisis.
Pengalaman bertahun-tahun dalam merawat dwarfisme membolehkan kami menganggap skim terapi berperingkat berikut adalah sesuai. Diagnosis dwarfisme pada pesakit dewasa biasanya tidak menimbulkan keraguan. Pada kanak-kanak kecil, jika gambaran klinikal tidak jelas, tempoh diagnostik diperlukan: 6-12 bulan di bawah pemerhatian tanpa terapi hormon. Pada masa ini, rawatan pengukuhan am yang kompleks ditetapkan; pemakanan yang mencukupi dengan peningkatan kandungan protein haiwan, sayur-sayuran dan buah-buahan dalam diet, vitamin A dan D, persediaan kalsium dan fosforus. Ketiadaan perubahan yang mencukupi dalam pertumbuhan dan perkembangan fizikal terhadap latar belakang ini dan pengesanan gangguan endokrin semasa peperiksaan adalah asas untuk memulakan terapi hormon.
Jenis utama terapi patogenetik untuk kerdil pituitari ialah penggunaan hormon pertumbuhan manusia, kerana kejadian kebanyakan kes dwarfisme sudah pasti bergantung pada satu atau lain bentuk kekurangannya. Oleh kerana kekhususan spesies hormon ini, hanya somatotropin manusia dan primat yang aktif untuk manusia. Ubat yang diasingkan daripada kelenjar pituitari orang yang mati akibat penyakit tidak berjangkit dan bukan neoplastik digunakan secara meluas di klinik. Somatotropin manusia diperolehi melalui sintesis bakteria menggunakan Escherichia coli oleh kejuruteraan genetik. Somatotropin manusia juga disintesis secara kimia, tetapi ia sangat mahal dan boleh dikatakan tidak digunakan di klinik. Pesakit dengan kekurangan hormon pertumbuhan endogen yang terbukti, dengan pembezaan rangka tidak melebihi tahap tipikal 13-14 tahun, dipilih untuk rawatan somatotropin. Tiada sekatan umur untuk rawatan.
Dos berkesan minimum yang boleh digunakan dalam tempoh pertama rawatan ialah 0.03-0.06 mg/kg berat badan. Dos yang paling berkesan ialah 2-4 mg 3 kali seminggu. Meningkatkan dos tunggal kepada 10 mg tidak disertai dengan peningkatan yang mencukupi dalam kesan pertumbuhan, tetapi menyebabkan pembentukan cepat antibodi kepada somatotropin.
Di negara kita, kerja mengkaji hormon pertumbuhan manusia telah dijalankan sejak tahun 1960. Dua rejimen rawatan telah diuji: berterusan dan terputus-putus dengan kursus 2-3 bulan dan selang yang sama antara mereka. Purata peningkatan ketinggian pesakit semasa tahun pertama rawatan ialah 9.52±0.39 cm, peningkatan berat badan ialah 4.4±0.14 kg. Dengan rawatan berterusan jangka panjang, purata peningkatan ketinggian ialah 0.82 cm/bulan, berat badan - 0.38 kg/bulan; dengan terputus-putus - 0.75 cm/bulan dan 0.4 kg/bulan, masing-masing. Rawatan berterusan memberikan peningkatan ketinggian yang lebih cepat dengan penurunan mendadak dalam kesan selepas 1-1.5 tahun, dengan rawatan terputus-putus, keberkesanannya dikekalkan selama 3-4 tahun, yang membolehkan kita mempertimbangkan rejimen rawatan kursus yang lebih sesuai. Penentuan tahap IGF-I (somatomedin C) boleh berfungsi sebagai penunjuk yang boleh dipercayai tentang kepekaan pesakit terhadap rawatan dengan ubat somatotropin. Peningkatan kandungan IGF-I selepas pengenalan hormon somatotropik membolehkan seseorang meramalkan kesan positif terapi. Kelebihan penting rawatan somatotropin ialah ketiadaan pecutan ossifikasi rangka terhadap latar belakangnya.
Cara yang paling penting untuk merawat dwarfisme ialah penggunaan steroid anabolik, yang merangsang pertumbuhan dengan meningkatkan sintesis protein dan meningkatkan tahap hormon somatotropik endogen. Rawatan dijalankan selama beberapa tahun, dengan penggantian beransur-ansur beberapa ubat dengan yang lain, daripada kurang aktif kepada sebatian yang lebih aktif. Perubahan dalam ubat anabolik ditunjukkan apabila kesan pertumbuhan berkurangan selepas 2-3 tahun, yang membawa kepada peningkatan tambahan dalam pertumbuhan. Rawatan dijalankan dalam kursus (tempoh rehat hendaklah separuh daripada tempoh rawatan). Dalam kes ketagihan, rehat yang lebih lama juga ditunjukkan (sehingga 4-6 bulan). Hanya satu daripada steroid anabolik ditetapkan pada satu masa. Menggabungkan 2 atau lebih ubat adalah tidak sesuai, kerana ini tidak meningkatkan kesan metabolik dan pertumbuhannya. Yang terakhir bergantung terutamanya pada umur pesakit dan tahap pembezaan tulang rangka pada permulaan rawatan. Kesan terbaik diperhatikan pada pesakit di bawah umur 16-18 tahun dengan osifikasi rangka tidak melebihi ciri tahap umur 14 tahun. Adalah dinasihatkan untuk memulakan rawatan sejurus selepas diagnosis, biasanya dari umur 5-7 tahun. Sebelum rawatan, adalah perlu untuk mengelak daripada menetapkan gonadotropin dan hormon seks, yang, sambil merangsang pertumbuhan, pada masa yang sama mempercepat pembezaan rangka. Prinsip dos steroid anabolik adalah daripada dos berkesan minimum kepada peningkatan secara beransur-ansur. Dos yang disyorkan untuk ubat yang paling biasa: nerobol (methandrostenol, dianabol) - 0.1-0.15 mg setiap 1 kg berat badan setiap hari secara lisan; nerobolil (durabolin) - 1 mg setiap 1 kg berat badan sebulan secara intramuskular, dos bulanan diberikan dalam 2-3 dos, masing-masing, selepas 15 atau 10 hari; retabolil (deca-durabolin) - 1 mg setiap 1 kg berat badan sebulan sekali secara intramuskular. Melebihi dos yang ditetapkan boleh menyebabkan androgenisasi. Dalam dos fisiologi, sebatian ini tidak memberi kesan ketara kepada keadaan alat kelamin dan pembezaan tulang rangka, yang membolehkan mereka digunakan untuk masa yang lama pada pesakit kedua-dua jantina. Kanak-kanak perempuan harus berada di bawah pengawasan pakar sakit puan, kerana dalam kes overdosis atau peningkatan sensitiviti individu, sesetengah pesakit mungkin mengalami tanda-tanda virilization, yang cepat mundur apabila rawatan dihentikan. Ubat oral yang dimetilasi kepada etilasi pada kedudukan ke-17 kadang-kadang boleh menyebabkan kesan kolestatik, oleh itu, dalam penyakit hati, keutamaan harus diberikan kepada sebatian anabolik parenteral, atau ubat oral harus digabungkan dengan agen koleretik. Sangat jarang, rawatan dengan steroid anabolik boleh menyebabkan reaksi alahan (gatal-gatal, ruam). Sekiranya tiada komplikasi, steroid anabolik digunakan selagi kesan pertumbuhan diperhatikan (sehingga 16-18 tahun, dan kadang-kadang lebih lama). Rawatan dijalankan dengan latar belakang terapi pengukuhan umum.
Sekiranya pesakit mempunyai tanda-tanda hipotiroidisme, ubat tiroid (tiroksin, tiroidin, thyrotom) ditetapkan serentak dalam dos yang dipilih secara individu.
Dalam rawatan kanak-kanak lelaki, langkah seterusnya ialah pentadbiran human chorionic gonadotropin. Ubat ini digunakan tidak lebih awal daripada 15-16 tahun, dan selalunya pada usia yang lebih tua untuk merangsang sel Leydig, yang mempercepatkan kedua-dua perkembangan dan pertumbuhan seksual (disebabkan oleh aktiviti anabolik androgen mereka sendiri). Dos 1000 hingga 1500 IU digunakan 1-2 kali seminggu secara intramuskular dalam kursus 2 bulan tidak lebih daripada 2-3 kali setahun. Sekiranya kesannya tidak lengkap, rawatan dengan human chorionic gonadotropin pada kanak-kanak lelaki berumur 16 tahun ke atas digantikan dengan pemberian dos kecil androgen (metiltestosteron pada dos 5-10 mg/hari sublingual).
Kanak-kanak perempuan berumur lebih 16 tahun boleh memulakan rawatan dengan dos kecil estrogen, menyerupai kitaran seksual biasa. Rawatan dijalankan selama 3 minggu setiap bulan, diikuti dengan rehat. Dalam fasa ke-2 kitaran, dari minggu ke-3, gonadotropin chorionic boleh ditetapkan pada dos 1000-1500 IU 3-5 kali seminggu atau ubat dengan kesan gestagen (pregnin, progesteron).
Peringkat akhir rawatan (selepas penutupan zon pertumbuhan) adalah pentadbiran berterusan dos terapeutik hormon seks yang sepadan dengan jantina pesakit, untuk mengembangkan sepenuhnya alat kelamin, ciri-ciri seksual sekunder, memastikan libido dan potensi seksual. Persediaan gabungan estrogen-progestogen (bukan ovlon, bisecurin, infekundin, rigevidon) adalah mudah untuk merawat pesakit wanita, dan persediaan androgen pelepasan berpanjangan (testenat, sustanon-250, omnadren-250) adalah mudah untuk merawat pesakit lelaki.
Rawatan pengukuhan am dijalankan (rejim, diet protein-sayuran, terapi vitamin, biostimulan). Penggunaan persediaan zink ditunjukkan, dalam mekanisme tindakan yang memainkan peranan utama dengan meningkatkan aktiviti IGF-1 (faktor pertumbuhan seperti insulin I).
Dengan kehadiran patologi organik dari sistem saraf pusat, terapi anti-radang, resorptif, dan dehidrasi diberikan. Terapi sistematik yang disasarkan memberikan kesan yang menggalakkan. Hasil daripada rawatan berperingkat jangka panjang, 148 (80.4%) daripada 175 pesakit dengan kerdil kedua-dua jantina berjaya mencapai ketinggian lebih 130 cm, 92 (52.5%) - lebih 140 cm, dan 32 (18.3%) - 150-160 cm atau lebih. Pada masa yang sama, 37 pesakit (21.2%) meningkatkan ketinggian mereka sebanyak 30 cm, 107 (61.1%) sebanyak 31-50 cm, dan 31 (17.7%) sebanyak 51-60 cm atau lebih.
Ramalan
Prognosis bergantung pada bentuk dwarfisme. Dalam jenis genetik dwarfisme, prognosis untuk kehidupan adalah baik. Dengan kehadiran tumor pituitari dan kerosakan organik pada sistem saraf pusat, ia ditentukan oleh dinamik perkembangan proses patologi utama. Kaedah terapi moden telah meningkatkan keupayaan fizikal dan kapasiti kerja pesakit dengan ketara, dan memanjangkan jangka hayat mereka. Semasa tempoh rawatan aktif, pesakit perlu diperiksa oleh doktor setiap 2-3 bulan, dengan terapi penyelenggaraan - setiap 6-12 bulan.
Pekerjaan pesakit yang sepadan dengan keupayaan intelek dan fizikal mereka adalah amat penting untuk penyesuaian sosial mereka.
Adalah dinasihatkan untuk memilih profesion yang tidak dikaitkan dengan usaha fizikal yang berat, tetapi membolehkan anda menunjukkan kebolehan intelektual, keupayaan untuk melakukan kerja yang tepat, dan bahasa.