Pakar perubatan artikel itu

Penerbitan baru
Neuroblastoma pada kanak-kanak: punca, diagnosis, rawatan
Ulasan terakhir: 04.07.2025

Semua kandungan iLive disemak secara perubatan atau fakta diperiksa untuk memastikan ketepatan faktual sebanyak mungkin.
Kami mempunyai garis panduan sumber yang ketat dan hanya memautkan ke tapak media yang bereputasi, institusi penyelidikan akademik dan, apabila mungkin, dikaji semula kajian secara medis. Perhatikan bahawa nombor dalam kurungan ([1], [2], dan lain-lain) boleh diklik pautan ke kajian ini.
Jika anda merasakan bahawa mana-mana kandungan kami tidak tepat, ketinggalan zaman, atau tidak dipersoalkan, sila pilih dan tekan Ctrl + Enter.
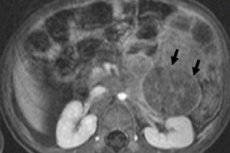
Dalam onkologi pediatrik, salah satu neoplasma ekstrakranial yang paling biasa adalah neuroblastoma pada kanak-kanak, yang merupakan tumor malignan embrionik neuroblast puncak saraf, iaitu, sel saraf embrio (tidak matang) sistem saraf simpatetik.
Epidemiologi
Menurut statistik dari International Neuroblastoma Risk Group (INRG), neuroblastoma menyumbang kira-kira 8% daripada semua penyakit onkologi pada kanak-kanak di seluruh dunia dan ketiga dalam kelaziman, selepas leukemia dan tumor otak.
Menurut data lain, neuroblastoma menyumbang kira-kira 28% daripada semua kanser pada bayi. Lebih daripada satu pertiga daripada kes neuroblastoma didiagnosis pada kanak-kanak di bawah umur satu tahun; umur purata diagnosis ialah 19-22 bulan. Lebih daripada 90% kes yang didiagnosis berlaku pada kanak-kanak berumur dua hingga lima tahun (dengan dominasi lelaki); insiden puncak diperhatikan pada usia dua hingga tiga tahun, dan kes pada kanak-kanak berumur lebih lima tahun menyumbang kurang daripada 10%.
Punca neuroblastoma
Dalam mengkaji punca neuroblastoma, penyelidik telah membuat kesimpulan bahawa tumor pada kanak-kanak ini berlaku kerana mutasi genetik sporadis semasa embriogenesis atau perkembangan awal selepas bersalin. Tetapi apa yang menyebabkan perubahan gen ini tidak diketahui, kerana tiada pengaruh faktor persekitaran teratogenik telah dikenalpasti.
Tumor ini boleh berlaku di mana-mana sahaja, termasuk mediastinum, leher, perut, kelenjar adrenal, buah pinggang, tulang belakang, dan pelvis.
Dalam kes yang jarang berlaku, neuroblastoma pada bayi mungkin dikaitkan dengan mutasi yang diwarisi. Khususnya, mutasi dalam gen protein membran CD246 pada kromosom 2 - enzim tyrosine kinase ALK, yang memastikan komunikasi antara sel dan memainkan peranan penting dalam fungsi sistem saraf; dalam gen protein PHOX2B (pada kromosom 4), yang terlibat dalam kematangan sel saraf.
Neuroblastoma juga mungkin dikaitkan dengan neurofibromatosis jenis 1 kanak-kanak,sindrom Beckwith-Wiedemann, dan hipoglikemia hiperinsulinemik (pankreatitis nesidioblastosis).
Faktor-faktor risiko
Hari ini, keturunan diiktiraf sebagai faktor risiko untuk perkembangan neuroblastoma pada kanak-kanak - kehadiran tumor ini dalam sejarah keluarga, serta anomali kongenital yang berkaitan dengan mutasi gen semasa perkembangan intrauterin. Ini terutama berlaku untuk kes-kes perkembangan beberapa neoplasma dalam organ yang berbeza.
Tiada faktor eksogen yang meningkatkan risiko tumor ini telah dikenal pasti oleh penyelidik.
Patogenesis
Mekanisme perkembangan neuroblastomas disebabkan oleh gangguan dalam pembezaan dan kematangan sel puncak neural - garisan sel dua hala yang terbentuk di tepi tiub neural dari lapisan kuman ektodermal embrio manusia. Sel-sel ini berhijrah (bergerak) dan membezakan kepada pelbagai jenis sel: neuron deria dan autonomi, sel neuroendokrin dan sel medula adrenal, sel rawan dan tulang kraniofasial, serta sel pigmen.
Dalam neuroblastoma, neuroblast yang berhijrah tidak matang, tetapi terus membesar dan membahagi, membentuk tumor. Dan patogenesis pembentukannya dikaitkan dengan mutasi gen berikut:
- dengan pertindihan sebahagian daripada urutan kromosom atau pertindihan segmen gen LMO1 pada kromosom 11, pengekodan protein RBTN1 dalam sel puncak neural embrio;
- dengan perubahan dalam nombor salinan gen NBPF10 pada kromosom 1q21.1, pengekodan protein DUF1220, yang mengawal percambahan sel stem saraf manusia. Gangguan ini membawa kepada sama ada pertindihan kromosom ini atau pemadamannya - ketiadaan sebahagian daripada DNA;
- dengan perubahan dalam gen penindas tumor ATRX (pada kromosom Xq21.1);
- dengan kehadiran salinan tambahan (penguatan) gen faktor transkripsi N-Myc pada kromosom 2, yang mengkodkan salah satu faktor transkripsi (protein pengikat DNA) yang mengawal aktiviti gen lain dan mengawal percambahan sel prekursor semasa pembentukan protein untuk pembentukan tisu dan organ janin. Penguatan gen ini mengubahnya menjadi onkogen, yang menimbulkan gangguan kitaran sel, peningkatan percambahan sel dan pembentukan tumor.
Gejala neuroblastoma
Tanda-tanda pertama neuroblastoma adalah tidak spesifik dan mungkin termasuk kehilangan selera makan (dan penurunan berat badan), keletihan semasa menyusu, demam, dan sakit sendi.
Gejala klinikal bergantung pada lokasi tumor utama dan kehadiran metastasis (yang berlaku dalam 60-73% kes).
Selalunya, neuroblastoma primer disetempat di medulla adrenal, yang mempunyai asal yang serupa dengan sel saraf. Pada kanak-kanak di bawah umur satu tahun, neuroblastoma adrenal didiagnosis dalam 35-40% kes. Simptomnya termasuk sakit perut, demam, penurunan berat badan, sakit tulang, anemia atau sindrom Pepper yang bersamaan: kerosakan hati yang meresap dengan hepatomegali yang teruk dan sindrom gangguan pernafasan.
Neuroblastoma retroperitoneal atau neuroblastoma retroperitoneal pada kanak-kanak, semasa ia membesar, mula menekan pundi kencing atau usus, yang boleh menyebabkan masalah dengan kencing atau buang air besar, bengkak kaki (pada kanak-kanak lelaki, skrotum membengkak).
Neuroblastoma mediastinum pada kanak-kanak (mediastinal neuroblastoma) sering menekan pada vena kava superior, dan ini boleh menyebabkan bengkak pada muka, leher, lengan, dan bahagian atas dada (dengan kulit menjadi merah kebiruan, dengan nodul subkutan). Batuk dan semput, masalah pernafasan (sesak nafas) atau masalah menelan (disfagia) muncul; nodus limfa yang diperbesarkan dicatatkan di leher, di atas tulang selangka, dan di ketiak.
Penyebaran sel tumor ke sumsum tulang membawa kepada anemia, trombositopenia dan leukopenia dengan kecenderungan untuk pendarahan.
Dan dengan metastasis di kawasan periorbital, lingkaran hitam atau lebam muncul di sekeliling mata. Tumor sebegini juga boleh menyebabkan sakit kepala dan pening, exophthalmia (bola mata membonjol), dan disebabkan oleh mampatan ujung saraf - kelopak mata terkulai (ptosis) dan pengurangan saiz murid (miosis).
Neuroblastoma perut atau neuroblastoma rongga perut pada kanak-kanak membawa kepada pembentukan anjing laut yang boleh dirasai di perut, kembungnya, kehilangan selera makan, sembelit, dan peningkatan tekanan darah. Tumor yang menekan pada saraf tunjang atau akar saraf boleh menyebabkan kebas dan kelemahan anggota badan, ketidakupayaan untuk berdiri, merangkak, atau berjalan. Jika tulang terjejas, sakit tulang mungkin berlaku.
Dalam kes tumor peringkat 3-4 dalam rongga perut dengan kerosakan nodus limfa, sel-sel tumor boleh memasuki parenkim buah pinggang, dan kemudian neuroblastoma buah pinggang yang meluas pada kanak-kanak berkembang, yang membawa kepada gangguan fungsinya.
Tahap
- Tahap 1 neuroblastoma adalah tumor utama yang disetempat dan diasingkan ke satu kawasan badan; nodus limfa pada kedua-dua belah pihak tidak terjejas.
- Neuroblastoma peringkat 2. Pada peringkat 2A, tumor primer terhad kepada satu kawasan tetapi besar; nodus limfa dua hala tidak terlibat. Pada peringkat 2B, nodus limfa di sisi badan di mana tumor terletak positif untuk metastasis.
- Neuroblastoma peringkat 3: tumor utama melintasi saraf tunjang atau garis tengah badan, metastasis unilateral atau dua hala ditemui dalam nodus limfa.
- Neuroblastoma peringkat 4: tumor telah merebak ke nodus limfa yang jauh, sumsum tulang, tulang, hati atau organ lain. Dan peringkat 4S ditentukan pada kanak-kanak di bawah umur satu tahun dengan tumor primer setempat, dengan penyebaran ke kulit, hati atau sumsum tulang.
Sistem Peringkat Risiko Neuroblastoma Antarabangsa (INRGSS)
INRGSS menggunakan faktor risiko yang ditentukan pengimejan (IDRF), yang merupakan faktor yang dilihat pada ujian pengimejan yang mungkin bermakna tumor akan menjadi lebih sukar untuk dibuang.
INRGSS membahagikan neuroblastoma kepada 4 peringkat:
- L1: Tumor tidak merebak dari tempat ia bermula dan tidak berkembang menjadi struktur penting. Ia terhad kepada satu bahagian badan, seperti leher, dada, atau perut.
- L2: Tumor tidak merebak (metastasis) jauh dari tempat ia bermula (contohnya, ia mungkin tumbuh dari sebelah kiri perut ke sebelah kiri dada), tetapi ia mempunyai sekurang-kurangnya satu IDRF.
- M: Tumor telah bermetastasis ke bahagian badan yang jauh (kecuali tumor pada peringkat MS).
- MS: Penyakit metastatik pada kanak-kanak di bawah umur 18 bulan, di mana kanser telah merebak hanya ke kulit, hati dan/atau sumsum tulang.
Komplikasi dan akibatnya
Neuroblastoma dicirikan oleh komplikasi dan akibat seperti:
- merebak (metastasis) ke nodus limfa, sumsum tulang, hati, kulit dan tulang;
- mampatan saraf tunjang (yang boleh menyebabkan kesakitan dan membawa kepada lumpuh);
- perkembangan sindrom paraneoplastik (disebabkan oleh tindakan bahan kimia tertentu yang dirembeskan oleh tumor, serta antigen disialoganglioside GD2 yang dinyatakan oleh sel-selnya), yang ditunjukkan oleh pergerakan mata sukarela yang cepat, koordinasi terjejas, kekejangan otot, dan cirit-birit;
- kambuh selepas selesai terapi utama (seperti yang ditunjukkan oleh amalan klinikal, neuroblastoma berisiko tinggi mempunyai kambuh semula dalam 50% kes).
Diagnostik neuroblastoma
Diagnosis neuroblastoma yang disyaki pada kanak-kanak memerlukan pemeriksaan, ujian makmal dan pengimejan.
Ujian darah dan air kencing diambil untuk katekolamin (norepinephrine dan dopamine) dan asid homovanillic atau vanillylmandelic (terbentuk semasa metabolisme hormon ini); ujian darah untuk enolase neurospecific, ujian imunosorben berkaitan enzim (ELISA) serum darah, dan analisis sumsum tulang (sampel yang diambil melalui tusukan aspirasi). Ujian DNA dilakukan untuk menentukan mutasi, dan biopsi dilakukan untuk pemeriksaan sitomorfologi tisu tumor.
Selepas sampel biopsi diambil, ia dihantar ke makmal di mana ia diperiksa di bawah mikroskop oleh ahli patologi (doktor yang mempunyai latihan khas dalam mengenal pasti sel-sel kanser). Ujian makmal khas juga sering dijalankan pada sampel untuk menunjukkan sama ada tumor adalah neuroblastoma.
Jika itu neuroblastoma, ujian makmal juga boleh membantu menentukan seberapa cepat tumor mungkin tumbuh atau merebak, serta rawatan yang mungkin berkesan.
Diagnostik instrumental menggambarkan neoplasma menggunakan ultrasound, X-ray, MRI atau CT, PET dengan pengenalan 18F-fluorodeoxyglucose atau pengimbasan MIBG - scintigraphy dengan metaiodobenzylguanidine. [ 1 ]
Diagnosis pembezaan
Diagnosis pembezaan termasuk ganglioneuroma jinak, ganglioneuroblastoma, rhabdomyosarcoma, nefroblastoma.
Siapa yang hendak dihubungi?
Rawatan neuroblastoma
Dalam neuroblastoma, rawatan bergantung kepada kumpulan risiko pesakit (peringkat proses tumor), penyetempatan tumor, ciri genomik sel tumor dan umur kanak-kanak. Dan ini mungkin termasuk pemantauan, pembedahan, kemoterapi, terapi sinaran, imunoterapi dan pemindahan sel stem hematopoietik.
Kemoterapi neoadjuvant atau adjuvant (sebelum atau selepas pembedahan) untuk neuroblastoma pada kanak-kanak, seperti mana-mana kemoterapi untuk kanser, diberikan dalam kursus: ubat diberikan selama beberapa hari berturut-turut, diikuti dengan rehat untuk badan pulih. Kitaran biasanya diulang setiap tiga hingga empat minggu.
Ubat-ubatan berikut (dan gabungannya) digunakan: Cyclophosphamide, Cisplatin atau Carboplatin, Doxorubicin (Adriamycin), Vincristine, Etoposide.
Kesan sampingan biasa ubat kemoterapi termasuk keguguran rambut, kehilangan selera makan, keletihan, loya dan muntah, ulser mulut, cirit-birit, atau sembelit. Kemoterapi boleh merosakkan sumsum tulang dan menyebabkan penurunan dalam bilangan sel darah.
Imunoterapi yang disasarkan (bertujuan kepada antigen tumor GD2) menggunakan ubat daripada kumpulan antibodi monoklonal (anti-GD2 MAb) Dinutuximab (Unituxin) dan Naxitamab. Mereka diberikan secara intravena dengan infusi yang berpanjangan, dalam kombinasi dengan faktor perangsang koloni granulosit-makrofaj (sitokin GM-CSF) dan interleukin-2.
Kesan sampingan ubat-ubatan ini termasuk sakit (selalunya sangat teruk), penurunan tekanan darah, peningkatan kadar jantung, sesak nafas (dengan kemungkinan pembengkakan saluran pernafasan), peningkatan suhu, loya, muntah dan cirit-birit, perubahan dalam komposisi selular dan mineral darah.
Untuk mengurangkan risiko kanser berulang selepas kemoterapi dos tinggi dan pemindahan sel stem, kanak-kanak dengan neuroblastoma berisiko tinggi dirawat dengan retinoid sistemik, asid 13-cis-retinoik (Isotretinoin). [ 2 ]
Rawatan pembedahan neuroblastoma - penyingkiran tumor, contohnya, adrenalektomi terbuka atau reseksi laparoskopi neuroblastoma adrenal; limfektomi (pembuangan nodus limfa yang terjejas), dsb. [ 3 ]
Untuk neuroblastoma berisiko tinggi, terapi sinaran boleh digunakan.[ 4 ]
Pencegahan
Memandangkan punca neuroblastoma pada kanak-kanak, satu-satunya langkah pencegahan mungkin adalah kaunseling genetik semasa merancang kehamilan. Tetapi perlu diingat bahawa tumor ini dikaitkan dengan mutasi yang diwarisi hanya dalam 1-2% kes.
Ramalan
Neuroblastoma infantil mempunyai keupayaan untuk mundur secara spontan.
Penanda prognostik
- Tumor berisiko tinggi, serta neuroblastoma pada kanak-kanak dari semua kumpulan umur dan semua peringkat (kecuali peringkat 4S) - dengan peningkatan ekspresi gen N-MYC dan penguatan onkogen N-Myc - mempunyai prognosis yang tidak menguntungkan yang menjejaskan jangka hayat.
- Mempunyai sel tumor yang kehilangan bahagian tertentu kromosom 1 atau 11 (dikenali sebagai pemadaman 1p atau 11q) mempunyai prognosis yang lebih teruk. Mempunyai bahagian tambahan kromosom 17 (keuntungan 17q) juga dikaitkan dengan prognosis yang lebih teruk.
- Sel neuroblastoma dengan jumlah DNA yang besar mempunyai prognosis yang lebih baik, terutamanya untuk kanak-kanak di bawah umur 2 tahun.
- Neuroblastoma yang mempunyai lebih banyak reseptor neurotropin, terutamanya reseptor faktor pertumbuhan saraf TrkA, mempunyai prognosis yang lebih baik.
Kelangsungan hidup oleh kumpulan risiko Childhood Oncology Group (COG).
- Kumpulan berisiko rendah: Kanak-kanak dalam kumpulan berisiko rendah mempunyai kadar survival 5 tahun lebih besar daripada 95%.
- Kumpulan risiko pertengahan: Kanak-kanak dalam kumpulan risiko pertengahan mempunyai kadar kelangsungan hidup 5 tahun 90% hingga 95%.
- Kumpulan berisiko tinggi: Kanak-kanak dalam kumpulan berisiko tinggi mempunyai kadar survival 5 tahun kira-kira 50%.
Kira-kira 15% daripada kematian kanser kanak-kanak adalah disebabkan oleh neuroblastoma. Peluang kelangsungan hidup jangka panjang untuk keganasan berisiko tinggi ini tidak lebih daripada 40%. Kadar kelangsungan hidup lima tahun keseluruhan ialah 67-74%, 43% dalam kumpulan umur satu hingga empat, dan lebih daripada 80% untuk neuroblastoma yang didiagnosis pada tahun pertama kehidupan.
Использованная литература