Pakar perubatan artikel itu

Penerbitan baru
Hamartoma
Ulasan terakhir: 29.06.2025

Semua kandungan iLive disemak secara perubatan atau fakta diperiksa untuk memastikan ketepatan faktual sebanyak mungkin.
Kami mempunyai garis panduan sumber yang ketat dan hanya memautkan ke tapak media yang bereputasi, institusi penyelidikan akademik dan, apabila mungkin, dikaji semula kajian secara medis. Perhatikan bahawa nombor dalam kurungan ([1], [2], dan lain-lain) boleh diklik pautan ke kajian ini.
Jika anda merasakan bahawa mana-mana kandungan kami tidak tepat, ketinggalan zaman, atau tidak dipersoalkan, sila pilih dan tekan Ctrl + Enter.
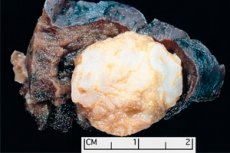
Pembentukan seperti tumor disetempat di mana-mana kawasan anatomi yang terhasil daripada pertumbuhan abnormal tisu jinak, dalam perubatan ditakrifkan sebagai hamartoma (dari bahasa Yunani hamartia - kesilapan, kecacatan). [ 1 ]
Epidemiologi
Secara statistik, hamartoma menyumbang 1.2% daripada neoplasma benigna. Kelaziman hamartoma pulmonari dianggarkan kira-kira 0.25% daripada populasi umum dan menyumbang sehingga 8% daripada semua neoplasma pulmonari. Kebanyakan hamartoma paru-paru didiagnosis secara kebetulan pada pesakit berumur 40 hingga 70 tahun, tetapi sangat jarang berlaku dalam amalan pediatrik.
Secara umum, kebanyakan hamartoma didiagnosis pada lelaki, walaupun di buah pinggang ia lebih biasa pada wanita dan dikenal pasti pada usia pertengahan.
Kira-kira 5% daripada tumor payudara benigna adalah hamartoma, dan ia paling kerap menyerang wanita berumur 35 tahun ke atas.
80-90% daripada lesi hamartoma pada otak dan lebih daripada 50% daripada hamartoma jantung dikaitkan dengan sklerosis tuberous.
Punca daripada hamartomas
Gamartoma tergolong dalam kecacatan kongenital dan merupakan pembentukan sifat jinak, yang terbentuk daripada tisu mesenkim yang berasal dari kepingan germinal. Dan punca kejadian mereka dikaitkan dengan pembahagian sel yang tidak terkawal dari tisu normal secara sitologi (penghubung, otot licin, lemak atau tulang rawan), ciri lokasi anatomi tertentu, dan pertumbuhan berlebihan fokus mereka semasa embriogenesis hampir mana-mana organ atau struktur anatomi.
Kejadian pelbagai hamartoma dalam pesakit yang sama sering dirujuk sebagai hamartomatosis atau hamartoma pleiotropik.
Tumor ini boleh berlaku secara sporadis atau dengan kehadiran penyakit warisan dominan autosomal tertentu serta sindrom yang ditentukan secara genetik.
Dalam banyak kes, hamartoma terbentuk apabila penyakit genetik yang jarang berlaku yang bersifat multisistemik - sklerosis tuberous - menjelma sejurus selepas kelahiran, atau dalam penyakit keluarga Recklinghausen - neurofibromatosis jenis 1. [ 2 ]
Faktor-faktor risiko
Faktor risiko utama untuk pembentukan hamartoma termasuk kehadiran sindrom genetik poliposis hamartomatous dalam sejarah pesakit, termasuk:
- Sindrom hamartoma berbilang - Sindrom Cowden, di mana pelbagai hamartoma asal ecto-, ento- dan mesodermal terbentuk, poliposis gastrousus dan manifestasi mukokutaneus diperhatikan;
- Sindrom Peutz-Jeghers-Turen (dicirikan oleh perkembangan polip hamartomatous jinak dalam saluran gastrousus);
- Sindrom Proteus;
- Sindrom Weil - poliposis juvana kolon;
- Sindrom Bannayan-Riley-Ruvalcaba, yang, seperti sindrom Cowden, menghasilkan pelbagai hamartoma (polip hamartomatous) usus;
- Sindrom Carney-Stratakis dan kompleks Carney.
Di samping itu, hamartoma terbentuk pada pesakit dengan sindrom Watson keturunan dan dalam kes-kes yang berlaku secara sporadis atau sindrom Pallister-Hall kongenital dengan hamartoma hipotalamus dan polydactyly.
Patogenesis
Mekanisme peningkatan percambahan tisu germinal dengan pembentukan kecacatan seperti tumor dalam pelbagai organ dijelaskan oleh penyimpangan kromosom dan mutasi gen yang boleh berlaku secara spontan atau diwarisi.
Dalam sklerosis tuberous, mutasi dalam gen TSC1 atau TSC2 - penekan tumor yang menghalang dan menghalang percambahan berlebihan - pertumbuhan dan pembahagian sel yang terlalu cepat atau tidak terkawal - telah dikenal pasti. Dan dalam neurofibromatosis jenis 1 dan sindrom Watson - mutasi germanium gen penindas tumor mitokondria NF1.
Dalam sindrom tumor hamartoma, yang menggabungkan sindrom Cowden, Protea, Bannayan-Riley-Ruvalcaba, dan poliposis juvana, patogenesis dikaitkan dengan mutasi gen PTEN, yang mengekod enzim yang terlibat dalam pengawalan percambahan dan dianggap sebagai gen penindas tumor.
Mutasi dalam gen STK11 mengekod struktur dan fungsi salah satu enzim serin transmembran, yang mengurangkan keupayaannya untuk menghalang pembahagian sel, membawa kepada sindrom Peutz-Jeghers-Turen, dengan perkembangan polip usus dan lesi kulit berpigmen. Mutasi dalam gen GLI3, faktor transkripsi yang terlibat dalam pembentukan tisu intrauterin, telah dikenal pasti dalam sindrom Pallister-Hall.
Oleh itu, pertumbuhan sel yang tidak terkawal akibat mutasi gen membawa kepada pembentukan hamartoma.
Gejala daripada hamartomas
Bergantung pada penyetempatan hamartoma, jenisnya dibezakan, dan masing-masing mempunyai struktur dan simptomologinya sendiri.
Hamartoma paru-paru
Hamartoma pulmonari boleh terbentuk di mana-mana bahagian lobus dan periferal paru-paru dan terdiri daripada tisu normal yang terdapat di dalam paru-paru: adiposa, epitelium, berserabut dan rawan. Dalam 80% kes, komponen kondroid (sel rawan hyaline) mendominasi dengan kemasukan adiposit - sel tisu adiposa dan sel epitelium saluran pernafasan. [ 3 ]
Nama-nama terdahulu: chondroid hamartoma, mesenchymoma, chondromatous hamartoma, atau hamartochondroma tidak disyorkan oleh WHO pada masa ini.
Hamartoma kistik mesenchymal paru-paru, sebaliknya, adalah kurang biasa dan dikaitkan dengan sindrom Cowden pada kebanyakan pesakit.
Lesi hamartomatous paru-paru mungkin tidak nyata, tetapi boleh menyebabkan gejala dalam bentuk batuk kronik (selalunya dengan hemoptisis), berdehit ketika bernafas dan kesukaran bernafas. [ 4 ]
Hamartoma jantung
Tumor jantung primer benigna pada orang dewasa termasuk hamartoma myocyte matang, dan pada bayi dan kanak-kanak dengan sklerosis tuberous, rhabdomyoma, iaitu, hamartoma miokardium ventrikel atau septum interventricular. [ 5 ]
Hamartoma kardiomiosit matang berkembang di dinding ventrikel (dan jarang di atria) dan mungkin muncul sebagai pelbagai lesi yang merupakan jisim padat yang berkait rapat dengan miokardium yang mendasari. Tumor boleh menyebabkan gejala kegagalan jantung: sakit dada, berdebar-debar dan aritmia, murmur jantung, edema, dyspnea, sianosis.
Rhabdomyomas jantung, yang kebanyakannya didiagnosis dalam tahun pertama kehidupan, terdiri daripada tisu otot jantung yang dibentuk oleh myoblasts embrionik dan mempunyai rupa jisim fokus pepejal tanpa kapsul.
Lazimnya, hamartoma ini hadir secara asimtomatik dan mundur secara spontan sebelum umur 4 tahun.
Lesi Hamartomatous juga dianggap oleh sesetengah pakar sebagai dikaitkan dengan myxoma kompleks Carney jantung. [ 6 ]
Gamartoma saluran gastrousus
Hamartoma gastrik ialah jisim mesenchymal dalam bentuk polip hiperplastik epitelium perut, polip Peutz-Jeghers dan hamartoma myoepithelial yang jarang berlaku - dengan berkas otot licin hipertrofi. Nama lain untuk hamartoma ini termasuk hamartoma mioglandular, hamartoma adenomyomatous dan adenomyoma gastrik. Manifestasi klinikal biasa termasuk dispepsia, sakit epigastrik, dan pendarahan GI atas. [ 7 ], [ 8 ]
Maklumat lanjut dalam bahan - Polyposis gastrik
Hamartoma usus ialah polip hamartomatous atau hiperplastik usus besar, yang didiagnosis sebagai adenoma adenomatous atau tubular. Apabila hamartoma disetempat di kelenjar Brunner duodenum, gejala ditunjukkan oleh rasa sakit di kawasan epigastrik; loya, muntah, dan kembung perut (menunjukkan halangan usus); dan, jika saiznya besar, pendarahan gastrousus. Dalam kes hamartoma myoepithelial ileum, pesakit mengadu sakit perut, berat badan berkurangan, dan mengalami anemia kronik. [ 9 ], [ 10 ]
Baca juga - Polip rektum
Hamartoma retrorectal ialah hamartoma sista atau sista berbilang ruang pada ruang retrorektal (tisu penghubung longgar antara rektum dan fasianya sendiri) yang paling biasa berlaku pada wanita pertengahan umur. Ia mempunyai rupa sista yang membonjol keluar dari dinding posterior rektum, yang dipenuhi dengan epitelium dan mengandungi gentian otot licin yang tersusun secara huru-hara. Hamartoma ini disertai dengan sakit perut bawah dan sembelit berulang. [ 11 ], [ 12 ]
Hamartomas hati dan limpa
Hamartoma bilier berbilang hati adalah hamartoma saluran hempedu intrahepatik interdollik yang dikaitkan dengan kecacatan perkembangannya semasa tempoh embrio. Hamartoma ini (tunggal atau berbilang) terdiri daripada kelompok saluran hempedu yang diluaskan secara sembarangan dan stroma fibrokolagen. [ 13 ]
Hamartoma biliari adalah tanpa gejala dan biasanya ditemui secara kebetulan (semasa pemeriksaan radiologi atau laparotomi). [ 14 ]
Neoplasma primer yang jarang dan sering dikesan secara kebetulan adalah hamartoma limpa, yang terdiri daripada unsur-unsur pulpa merah limpa - dalam bentuk jisim homogen yang jelas dengan konsistensi teguh. Kecacatan ini boleh tunggal atau berbilang; apabila memerah parenkim splenik, mungkin terdapat rasa tidak selesa dan sakit di kawasan subcostal kiri. [ 15 ], [ 16 ]
Hamartoma buah pinggang
Hamartoma buah pinggang yang paling biasa didiagnosis sebagai angiomiolipoma buah pinggang, kerana tumor jinak ini terdiri daripada tisu adiposa matang dengan gentian otot licin dan saluran darah yang tertanam. Ia terbentuk dalam sklerosis tuberous dalam 40-80% kes. Meningkatkan saiz hamartoma (lebih daripada 4-5 cm) membawa kepada kesakitan dan kemunculan darah dalam air kencing. [ 17 ], [ 18 ]
Hamartoma payudara
Definisi diagnostik hamartoma payudara yang diterima WHO ialah istilah seperti adenolipoma, kondrolipoma dan myoid hamartoma. Walaupun sering dipanggil fibroadenolipoma oleh ahli mammologi, kerana pembentukan tumor mengandungi sel-sel berserabut, kelenjar, dan tisu adiposa yang tertutup dalam kapsul tisu penghubung nipis dengan garis besar yang berbeza. Kalsifikasi fokus boleh diperhatikan pada visualisasi. Dalam kes ini, manifestasi klinikal tidak hadir. [ 19 ], [ 20 ]
Baca juga - Tumor payudara
Hamartoma otak
Satu pertiga daripada pesakit dengan tuberous sclerosis mempunyai hamartoma otak dalam bentuk pertumbuhan kortikal intrakranial atau tuberkel dalam pelbagai lobus - di sempadan jirim kelabu dan putih - atau nodul subependymal di sepanjang dinding ventrikel otak. Hamartoma astrocytic, astrocytoma sel gergasi subependymal dengan gangguan kortikal, neuron dismorfik dan sel glial besar parenkim otak (astrocytes), juga boleh terbentuk. Gejala hamartoma serebrum termasuk serangan sawan dan terencat akal pada kanak-kanak. [ 21 ], [ 22 ]
Kecacatan yang jarang berlaku yang berlaku semasa embriogenesis dan hadir semasa lahir ialah hamartoma hipotalamus, yang merupakan jisim neuron heterotopik dan sel glial. Apabila otak kanak-kanak membesar, tumor membesar tetapi tidak merebak ke kawasan serebrum yang lain. [ 23 ], [ 24 ]
Jika tisu hipertrofi terbentuk di bahagian anterior hipotalamus (ubi cinereum), di mana kelenjar pituitari melekat padanya, kecacatan menunjukkan gejala perkembangan seksual pramatang pusat (sebelum umur 8-9 tahun): penampilan ruam jerawat, perkembangan awal kelenjar susu dan menarche awal pada kanak-kanak perempuan; rambut kemaluan awal dan mutasi suara pada kanak-kanak lelaki.
Apabila hamartoma terbentuk di bahagian posterior hipotalamus, mungkin terdapat keabnormalan dalam aktiviti elektrik otak, yang pada awal bayi dimanifestasikan oleh sawan, dan pada peringkat kemudian (umur 4 hingga 7 tahun) oleh epilepsi dengan sawan epileptik fokus dengan ketawa secara tiba-tiba atau dengan tangisan yang tidak disengajakan, sawan atonic dan juga tonik-klonik, serta sawan ingatan. masalah kognitif.
Hamartoma pituitari ialah adenoma pituitari jinak yang berlaku secara sporadis.
Orang dewasa pertengahan umur dengan sindrom Cowden mungkin mempunyai jisim seperti tumor yang jarang berlaku, hamartoma otak kecil, didiagnosis sebagai gangliocytoma cerebellar displastik atau penyakit Lhermitte-Duclos. Gejala mungkin tidak hadir atau nyata sebagai sakit kepala, pening, gangguan koordinasi pergerakan, dan lumpuh saraf kranial individu.
Hamartoma nodus limfa
Apabila sel-sel otot licin dan tisu adiposa, serta saluran darah dan stroma kolagen nodus limfa inguinal, retroperitoneal, submandibular dan serviks berlebihan, hamartoma angiomyomatous nodus limfa atau nodular angiomyomatous hamartoma terbentuk - dengan penggantian separa atau lengkap parenkimnya. [ 25 ], [ 26 ]
Hamartoma pada kulit
Dengan kehadiran sklerosis tuberous atau neurofibromatosis, pelbagai hamartoma kulit diperhatikan, paling kerap dalam bentuk bintik-bintik hipopigmentasi; bintik kopi dan susu; angiofibroma (di pipi, dagu, lipatan nasolabial); bintik-bintik shagreen pelbagai penyetempatan (iaitu nevi tisu penghubung); plak berserabut pada dahi, kulit kepala atau leher.
Manifestasi dermatologi yang jarang berlaku bagi sklerosis ubi (terutamanya pada lelaki) ialah folikulokistik dan hamartoma kolagen, dicirikan oleh pemendapan kolagen yang banyak dalam dermis, fibrosis perifolikular sepusat, dan sista subkutan berbentuk corong berisi keratin yang dilihat pada pemeriksaan histopatologi. [ 27 ]
Kepada hamartoma yang terdiri daripada melanosit (sel yang menghasilkan pigmen melanin), kebanyakan pakar juga merujuk kepada pelbagai neoplasma melanocytic, khususnya nevi melanocytic kongenital, yang mewakili kelainan embriogenesis.
Dari segi etiologi, hamartoma yang terdiri daripada tisu vaskular juga merupakan hemangioma pada kulit.
Pesakit dengan sindrom Peutz-Jeghers-Thuren mempunyai hamartoma dalam bentuk pigmentasi bertompok pada kulit dan membran mukus - lentiginosis periorificialis
Kes-kes hamartoma ektodermal-mesodermal papular linear (Hamartoma moniliformis) menunjukkan ruam papular berwarna daging linear pada kepala, leher, dan bahagian atas dada.
Dan hamartoma sebocytic adalah hamartoma kelenjar sebum, baca lebih lanjut dalam penerbitan - nevus sebum.
Hamartoma mata
Lesi hamartomatous berpigmen pada iris dalam neurofibromatosis jenis 1 dan sindrom Watson - dalam bentuk kelompok nodular melanosit dendritik - ditakrifkan sebagai hamartoma iris atau nodul Lisch. Mereka adalah telus (biasanya tidak menjejaskan penglihatan) bulat berbentuk kubah kuning-coklat papula yang menonjol di atas permukaan iris.
Dan pesakit dengan angiofibroma juvana nasofaring dan poliposis adenomatous familial sering membina hamartoma gabungan retina dan epitelium pigmen retina - dalam bentuk bintik hitam pada bahagian tengah (makular) retina. [ 28 ]
Hamartoma hidung
Nasal hamartoma ditakrifkan oleh pakar sebagai nasal chondromesenchymal hamartoma atau nasal chondroma, disebabkan oleh pembiakan jinak epitelium pernafasan, kelenjar submukosa dan mesenkim tulang kondro. Manifestasi klinikalnya bergantung pada saiz dan penyetempatan lesi dan termasuk: hidung tersumbat, kesukaran bernafas hidung dan penyusuan pada bayi, lelehan hidung berair jernih, dan pendarahan hidung. Hamartoma boleh tumbuh bersama kanak-kanak dan merebak ke orbit mata, mengakibatkan anjakan bola mata, strabismus atau okulomotor ke hadapan atau ke belakang. [ 29 ]
Hamartoma pada kanak-kanak
Semua lesi hamartomatous yang disebutkan di atas pelbagai organ dan struktur anatomi terdapat pada kanak-kanak dengan sindrom yang sepadan.
Bayi yang baru lahir mengalami hamartoma mesenchymal dinding dada atau hamartoma cartilaginous rusuk, yang merupakan jisim pepejal tidak bergerak yang terhasil daripada pertumbuhan berlebihan fokus unsur rangka normal dengan unsur cartilaginous, vaskular dan mesenchymal. Hamartoma ini boleh menyebabkan kegagalan pernafasan dan perkembangan sindrom gangguan pernafasan. Hamartoma mesenchymal hati adalah tumor hati benigna kedua paling kerap pada kanak-kanak. Pembentukan seperti tumor ini (lebih kerap dilokalkan di lobus kanan organ) terdiri daripada sel stroma mesenchymal, hepatosit dan sel epitelium lapisan saluran hempedu. Gambar klinikal termasuk jisim yang boleh dirasai dalam rongga perut, anoreksia dan penurunan berat badan, dan dalam kes saiz yang ketara (sehingga 10 cm dan lebih) tumor meliputi saluran hempedu ekstrahepatik dan vena kava inferior, yang membawa kepada jaundis dan edema pada bahagian bawah kaki.
Hamartoma ialah nefroma mesoblastik kongenital (berlaku dalam 1 dalam 200,000 bayi) yang boleh mengakibatkan kembung perut pada bayi baru lahir dengan jisim konsistensi padat yang boleh dirasai di kuadran kanan atas abdomen. Bayi juga mungkin mengalami pernafasan cetek yang cepat.
Anomali kongenital yang jarang berlaku termasuk hamartoma berserabut pada bayi, yang berlaku pada kanak-kanak dalam dua tahun pertama kehidupan dan muncul sebagai jisim nodular yang tidak menyakitkan dalam tisu subkutan pada axilla, leher, bahu dan lengan bawah, belakang dan dada, paha, kaki, dan alat kelamin luar.
Hamartoma angiomatous eccrine pada kanak-kanak mungkin hadir semasa lahir atau nyata pada awal kanak-kanak. Tumor jinak yang bersifat hamartomatous ini biasanya mempunyai rupa nodul kebiruan atau keperangan dan/atau plak yang terhasil daripada percambahan tisu kelenjar peluh ekrin dan kapilari di lapisan tengah dan dalam dermis. Hamartoma ini boleh menyebabkan hiperhidrosis setempat dan peningkatan pertumbuhan rambut.
Komplikasi dan akibatnya
Umumnya dipersetujui bahawa hamartoma jarang berulang atau berubah menjadi tumor malignan. Mereka sering menunjukkan sedikit atau tiada simptom dan kadangkala hilang dari semasa ke semasa. Tetapi dalam kes yang lebih teruk dan bergantung pada tapak pembentukan, kecacatan ini boleh menyebabkan komplikasi dan akibat yang serius.
Pertama sekali, hamartoma boleh membesar sehingga saiznya mula menekan pada tisu dan organ sekeliling, mengganggu fungsinya.
Hamartoma jantung pada kanak-kanak boleh menyebabkan keabnormalan irama jantung yang berterusan, kecacatan injap, dan gangguan aliran darah intrakardiak dengan kegagalan jantung kongestif berikutnya.
Komplikasi polip hamartomatous saluran GI adalah pendarahan gastrousus, halangan dan intususepsi usus (dengan kemungkinan membawa maut). Dan hamartoma buah pinggang yang besar boleh mencetuskan pecahnya buah pinggang.
Hamartoma dalam otak boleh menyebabkan sindrom hidrosefalus obstruktif.
Dalam hamartoma hipotalamus dan pituitari, pengeluaran hormon somatotropik (hormon pertumbuhan) mungkin terjejas, yang membawa kepada perkembangan nanisme hipofisis (hypopituitarism) pada kanak-kanak. Hamartoma hipotalamus pada kanak-kanak juga boleh menyebabkan epilepsi tahan dadah.
Komplikasi hamartoma epitelium pigmen retina penuh dengan disfungsi retina dan/atau saraf optik, edema makula, neovaskularisasi koroid, dan detasmen retina.
Diagnostik daripada hamartomas
Bahagian penting dalam diagnosis hamartoma dan sindrom yang berkaitan ialah pengumpulan anamnesis, termasuk sejarah keluarga.
Ujian makmal termasuk ujian darah: klinikal am; elektrolit serum; profil limfosit; tahap kalsium, kalium, fosfat dan urea; dan ujian fungsi hati. Jika boleh, biopsi tusukan aspirasi jarum halus pada jisim dilakukan, kerana pemeriksaan histologi adalah penting dalam diagnosis dan pilihan taktik rawatan.
Diagnostik instrumental menyediakan visualisasi pembentukan seperti tumor hamartomatous dan pengenalpastian penyetempatannya yang tepat, yang mana X-ray, angiography, electroencephalography (EEG), ultrasound (sonografi), CT (computed tomography), PET (positron emission tomography), MRI (magnetic resonance imaging) digunakan.
Diagnosis pembezaan
Dalam mana-mana jisim yang tidak normal, diagnosis pembezaan adalah sangat penting. Oleh itu, tuberkuloma dan hamartoma dibezakan; hamartoma pulmonari dan kanser paru-paru primer, karsinoid bronkogenik, penyakit metastatik. Hamartoma otak harus dibezakan daripada craniopharyngioma dan glioma hypothalamic-chiasmatic. Dan diagnosis pembezaan hamartoma sebagai nefroma mesoblastik kongenital termasuk tumor Wilms (nephroblastoma malignan), sarkoma sel jernih buah pinggang dan tumor buah pinggang yang mengeras pada bayi.
Siapa yang hendak dihubungi?
Rawatan daripada hamartomas
Jika hamartoma adalah asimtomatik dan ditemui secara tidak sengaja, tiada rawatan diperlukan, tetapi perlu untuk memantau "tingkah laku" dan keadaan pesakit. Dalam kes lain, terapi bertujuan untuk mengurangkan keamatan gejala dan mencegah komplikasi. Sebagai contoh, dalam hamartoma hipotalamus dengan gejala akil baligh pramatang, ubat-ubatan tertentu yang menghalang pembebasan hormon tertentu ditetapkan. Ubat jantung digunakan untuk merawat gejala kegagalan jantung pada pesakit dengan hamartoma jantung.
Pembuangan hamartoma secara pembedahan ditunjukkan untuk mengesahkan diagnosis dan dalam kes gejala sengit yang tidak boleh dibetulkan secara perubatan.
Sebagai contoh, hamartoma paru-paru boleh direseksi dengan pemotongan baji dan, dalam kes yang teruk, dengan penyingkiran lobus paru-paru (lobectomy). Hamartoma payudara juga boleh dipotong, dan jika ia besar, mastektomi separa atau lengkap mungkin diperlukan.
Termoablasi frekuensi radio stereotaktik atau ablasi laser boleh digunakan untuk membuang polip hamartomatous. Radiosurgeri dengan sinar gamma sangat fokus - pisau gamma untuk hamartoma hipotalamus atau hamartoma astrocytic - juga digunakan.
Pencegahan
Satu-satunya kaedah untuk mencegah perkembangan hamartoma boleh dianggap sebagai pemeriksaan genetik ibu bapa masa depan kanak-kanak itu.
Ramalan
Prognosis keseluruhan anomali kongenital ini bergantung pada penyetempatan dan saiz neoplasma, serta pada komorbiditi dan kesihatan umum pesakit.